Magnesium Research (1993) 6, 2, 167-177
Regulation of sodium and potassium pathways by magnesium in
cell membranes
Michel Bara1, Andrée Guiet-Bara1
and Jean Durlach2
1Biology of Reproduction, Université P.M.
Curie, quai Saint-Bernard, Paris, France; 2SDRM,
Hôpital Saint-Vincent de Paul, rue Denfert Rochereau,
Paris, France
Summary: Magnesium plays an
important role in a large number of cellular processes by acting
as a cofactor in enzymatic reactions and transmembrane ion
movements. Magnesium is a modulator of Na,K ion transport systems
in numerous tissues. In this study, the interactions between
magnesium and Na,K pathways are described. In the paracellular
pathway, Na,K transports are generally increased by
Mgo. In the cellular pathway, there are various
processes: (1) Potassium channels - Mgi
blocks the outward currents, first by interfering with the
passage of K+ ions and inducing rectification of the
channel current-voltage relationship, and secondly by completely
blocking the channel pore and reducing the channel open
probability; Mgo increases the K+ channel
permeability in a leaky membrane. (2) Sodium channels
Mgi blocks outward currents in a voltage- and
dose-dependent manner, acts as a fast blocker by screening of
surface charges, and produces an open channel block in several
Na+ channels; Mgo increases Na+
transport in toad bladder and human amnion at high concentration
by acting on the driving force of the sodium pump. (3) Na/K
pump - Mgi and Mgo stimulate the Na/K
exchange at low concentration and inhibit it at high
concentration, by a stabilization of E2 forms of the enzyme which
would reduce the rate of turnover of the pump. (4) Na-K-Cl
cotransport increasing Mgo concentration
stimulates this system in red cells and human amnion, and the
bumetadine-sensitive K+ transport is sensitive to
Mgi (5) KCl cotransport - The increase in
Mgi inhibits this cotransport. (6) Na-H
antiport - Na/H exchange responds to manipulations of cell
magnesium but the effect is probably not a direct one; magnesium
is required not for the transport process per se, but
for the transduction of the volume stimulus (7) H-K pump
- Mg activates this system. (8) Na-Ca antiport - The
activity of this antiporter is inhibited by Mgo; the
inhibition by magnesium is competitive with calcium. (9)
Na-Mg exchange - in this system, the Na+
gradient provides the energy for net Mg2+-extrusion.
In conclusion, intracellular and extracellular magnesium may be
an important physiological regulator of the sodium and potassium
pathways in the cell.
Key words: Cell membranes,
extracellular magnesium, intracellular magnesium, pathways,
potassium, sodium.
Introduction
Magnesium, one of the most abundant intracellular divalent
cations, plays an important role in a large number of cellular
processes by regulating various biochemical reactions and acting
as a cofactor in transmembrane movements 1. Magnesium
has profound effects on solute and water transport in various
cells, and intracellular Mg2 is known to interact with
several channels. Excess Mg2 in the extracellular
fluid is known, for example, to depress synaptic transmission and
to cause rectification of the ionic channels coupled to
N-methyl-D-aspartate receptor 2. When considering the
transport systems, it is important to differentiate intracellular
magnesium (Mgi) and extracellular magnesium
(Mgo).
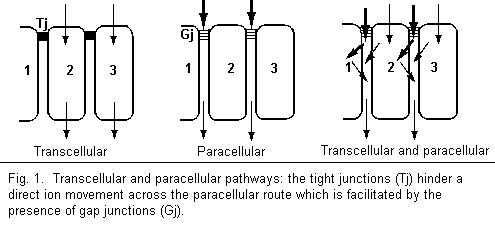
The action of magnesium depends on the cell membrane category.
It is important to distinguish the tight cell membranes (presence
of tight junctions) from the leaky cell membranes (presence of
desmosomes and gap junctions). In cell membranes, ionic movements
are carried out via two pathways: indeed, the paracellular
pathway is hindered by the presence of tight junctions, ionic
movements being first cellular and then paracellular. In a leaky
cell membrane, the ionic movement may be either directly
paracellular, or cellular, or cellular and paracellular (Fig
1).
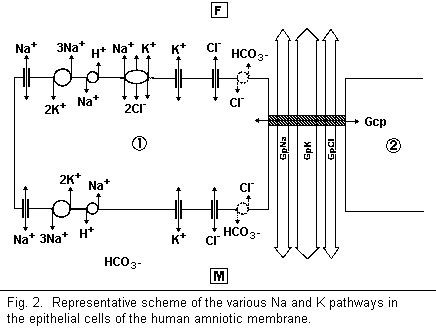
In this review, the interactions between magnesium and the
sodium and potassium pathways in cell membranes are examined. The
most important pathways, observed by the use of inhibitors, are
the following: paracellular Na (inhibited by
triaminopyrimidinium) and K (protamines) pathways, cellular
Na+ channels (chloride), cellular K+
channels (Ba2+) , Na/K-ATPase (ouabain), Na/H antiport
(amiloride), K/Cl cotransport, NaK-Cl cotransport (bumetanide),
K/H pump, Na/Ca antiporter, and the Na/Mg exchange (inhibited by
quinidine and Mn) (Fig. 2).
Methods
There are various methods of studying the interactions between
Mg and Na,K pathways.
(1) The patch-clamp method
Recordings of single channel currents 3 were
performed using heat polished patch electrodes. The experiments
utilized either the cell-attached 'open cell' recording
configuration or excised 'inside-out'and'outside-out'membrane
patches (in these cases the concentration of internal or external
magnesium may be easily modified).
(2) The voltage-clamp method (recording of total
channels)
There are several methods, particularly the classical oil gap
voltage-clamp method 4 and the whole cell
configuration of the patch-clamp techniques.
(3) Paracellular and cellular conductance
measurements
Conductance measurements are achieved after implantation of
two microelectrodes in the cell, passage of current, and
recording the membrane potential 5.
(4) Isotopic measurements
Measurement of net and unidirectional isotopic sodium fluxes,
for example during short-circuit conditions, were performed with
22Na and 24Na according to the method of Walser 6. In
order to define the site of action of magnesium, experiments were
conducted with a protocol in which the driving force of the
sodium pump, as well as the conductances of the active transport
pathway and the passive shunt pathway, were measured by the
method of Siegel & Civan 7 in the presence and
absence of magnesium in the mucosal solution,
(5) Use of Ionophores
Ionophores such as A23187 rapidly transport magnesium across
the cell membrane and modify intracellular magnesium
concentration.
Interactions between magnesium and the paracellular Na,K
pathways
The major part of the studies concerns the effects of
extracellular magnesium. Extracellular Mg2+ blocks the
passive movements of Na+ and K+ across the
membrane of heart cells 8. An attractive explanation
for the inhibition of transport is that Mg ions bind to and thus
screen fixed negative charges on cell membranes. In the toad
bladder, Mg increases the net sodium transport 9-10
but decreases it in rat renal proximal tubule and in jejunum.
In a leaky membrane (human amniotic membrane), there are
various possibilities concerning the ionic transfer across the
intercellular space (Figs. 3, 4): (1) a direct pathway across
intercellular space, via desmosomes and gap junctions, the ionic
mobility being a function of the size of the hydrated or
dehydrated ion; (2) a passage in the intercellular space, then
via the gap junction in the adjacent cell, this pathway being
regulated by the electrical coupling between two adjacent cells;
(3) a passage in the intercellular space, then via the surface
charges in the adjacent cell, this pathway being a function of
the distribution and organization of the surface charges of the
bilayer unit. This organization implicates a biphasic effect of
magnesium on sodium transport: at oral concentrations the
transfer is decreased because of screening of the surface
charges; at parenteral concentrations, it is increased because
magnesium ions release the surface charges which become
accessible to sodium ions 11-13.
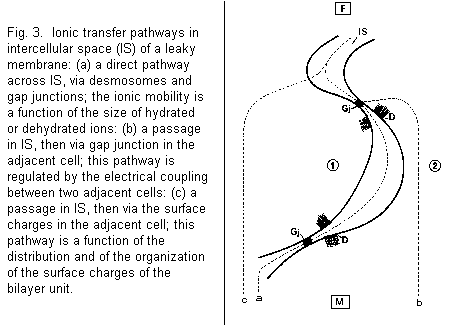
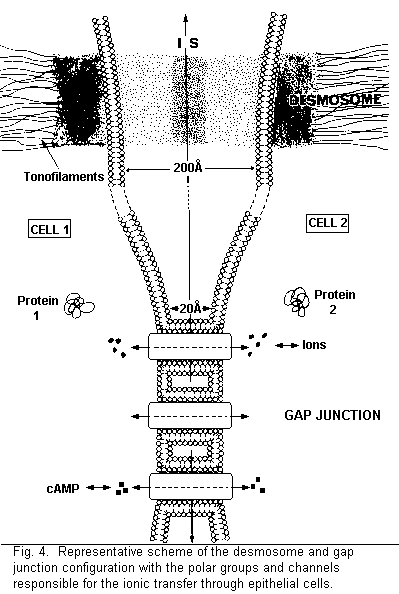
Intracellular Mg2+ would be without effect on the
passive influx (backleak) component 9.
Interactions between magnesium and cellular Na,K
pathways
Na+ channels
The decrease of the Na+ channel conductance of the
oocyte, at high potentials, can be explained by a blocking action
of Mgi in a voltage- and dose-dependent manner. The
reduction in Na+ conductance for outward currents is
caused by Mg block. Pusch 14 showed that
Mgi acts as a fast blocker rather than gradually
decreasing current, e.g. by screening of surface
charges. The screening of negative surface charges at the
intracellular side of the membrane has been proposed as an
alternative mechanism for the blocking effect of Mgi
15. In 1991, Lin et al. l6,
studying cerebellar granule cells, proposed a model in which
Na+ and Mg2+ competitively occupy the
sodium channel. In this model, Na+ and Mg2+
have to bind to the sodium channel during their passage through
the membrane. The blockade of sodium currents may be caused by
magnesium occupying the sodium channel, which prevents the
further binding of sodium (if the binding site for
Mg2+ is one of binding sites for Na+ during
its passage) or interferes with the normal accommodation of
Na+ by sodium channels. Blockade of the sodium channel
by Mgi may occur for two different reasons. The first
possibility is related to the selectivity of sodium channels.
Mgi enters the sodium channels and binds to an inside
site during the membrane depolarization. Because of the
selectivity of the pore (the permeability ratio of
Mg2+ to Na+ through the sodium channel is
less than 0.1 17), Mg2+ is not further
permeable and is even re-extruded due to the small extracellular
Na+ inward flux through the channel. The other
possibility is that the strong binding force between
Mg2+ and the binding site retards the release of
Mg2+ from the channel. In fact, the dissociation
constant value for Mg2+ binding within the sodium
channel at zero membrane potential is 8.65 ± 1.51 mM,
which is smaller than that for Nai (83.76 ±
7.60 mM). Most probably, these two possibilities may coexist and
influence each other, which may lead to the entrance of
Mg2+ into the channel several times during the
depolarization, giving high frequency flickering at the single
channel level and thus producing an apparently single conductance
decrease.
The study of external divalent cations shows that the order of
efficacy for the gating shift at a concentration of 5 mM (Ca >
Ba > Sr > Mg) was similar, but not identical, to that for
block (Ca > Mg > Sr > Ba). Thus, the binding sites for
external divalent cations associated with the gating shift are
probably somewhat different from those associated with block.
This tends to argue against the possibility that occupancy of the
divalent cation blocking site in the channel pore causes the
depolarization gating shift 18. This suggests that a
negative charge is normally exposed to the external side of the
channel in the closed state and that when the channel opens, this
group moves into the channel protein. In a leaky epithelium
(human amniotic membrane), Mgo, (2 mM) increases the
Na+ movement in the sodium channels. This is due to an
interaction with the negative surface charges by a binding
effects 19 (Fig. 5).
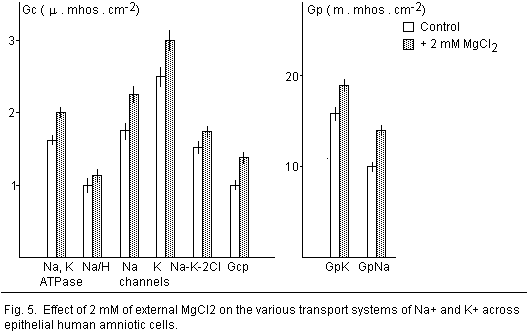
K+ channels
Inhibitory effects of intracellular Mg2+ have been
reported for outward currents through several channels.
(1) Muscarinic K+ channel
Stimulation of the muscarinic acetylcholine receptor causes an
increase in outward current by activating a class of
K+ channel in cardiac cells, thereby slowing the
pacemaker activity. Hory & Irisawa 20,21, studying
guinea pig atrial cells, showed that depletion of internal
Mg2+ increased outward muscarinic K+
currents but decreased inward currents, thereby reducing the
inwardly rectifying property of the channels. Mgi acts
as a 'fast' ionic blocker by plugging the open channel pore from
the inside of the cell and prevents outward K+ passage
22, thereby exhibiting an inward rectifying property.
Intracellular Mg2+. at physiological concentration,
has a dual action on the muscarinic K+ channel: first,
Mg2+ activates the channel in the presence of
guanosine triphosphate (GTP) through GTP binding proteins (G
proteins), and secondly, it blocks outward currents through the
channel causing the inwardly rectifying properties.
(2) Inwardly rectifying K+ channel
The inwardly rectifying K+ channel provides the
resting K conductance in a variety of cells. This channel acts as
a valve or diode, permitting entry of K+ under
hyperpolarization, but not its exit under depolarization. The
channel conductance is ohmic and the well known inward
rectification of the resting K conductance is caused by rapid
closure of the channel accompanied by a voltage-dependent block
by intracellular Mg2+ ions at physiological
concentrations 23 24. The outward single channel
currents through the inwardly rectifying K+ channel
responsible for the resting conductance appeared after the
intracellular side of the membrane was exposed to an artificial
solution. The outward current is blocked by internal
Mg2+ at physiological concentrations. The outward open
channel was found to fluctuate between sublevels in the presence
of Mg2+ at micromolar concentrations. Binomial
distribution of occupancy at each current level suggests that the
inwardly rectifying K+ channel is composed of three
identical K+conducting subunits 25,27 which
operate cooperatively to form a single channel, each of which is
blocked by internal Mg2+ in a voltage-dependent manner
28.
(3) ATP-sensitive K+ channel
Potassium-selective single membrane channels, inhibited by
adenosine triphosphate (ATP), applied to the internal surface of
the cell membrane (K+ -ATP channels) have been
described in cardiac and muscle cells 29,30 and in
insulin-secreting cell 31.
The rectification of currents passing through the
K+ -ATP channel resulted largely from the blockade of
the outward movement of K+ ions through the channel
pore by Mg2+ ions upon the internal surface
32,33, thereby resulting in a negative slope of the
conductance at positive potentials.
(4) Ca2+-activated K+
channel
Internal Mg2+ reversibly decreases the
K+ current in a concentration-dependent manner in the
large conductance Ca2+-activated K+ channel
in primary cultures of rat skeletal muscle 35.
Mgi2+ and K+ appear to compete
and the apparent competition may arise from the screening of
negative charges at the inner surface of the channel protein. The
blocking effects of Mg2+would be present under
physiological concentrations of this ion.
There are many other K+ channels regulated by
Mg2+ ions. For example, external modifications of the
Mg2+concentration indicated that Mg2+ ions
are activators of the K+ channels on the maternal and
fetal sides of the human amniotic membrane 36 (Fig.
5).
Na/K-ATPase pump
Na/K-ATPase is a membrane-bound enzyme responsible for the
active transport of K+ and Na+ across the
plasma membrane which maintains the ionic gradients. It consists
of two subunits, the catalytic subunit and the β subunit,
which is a glycoprotein. During ion transport, the enzyme is
believed to proceed through two major conformational states, El
and E2, characterized by different affinity for K+ and
Na+. Within the framework of the two conformational
states, the enzyme passes through different substrates, in the
formation of which divalent cations can play an important role
either by binding to the protein sites or, indirectly, through
interaction with phospholipids 37. Gupte et
al. 38 suggested that Mg2+ interacts
primarily with the polar headgroups of the phospholipid in
purified Na/K-ATPase, inducing structural changes in the lipid
vesicles. Amler et al. 39 showed that
Mg2+, increased the order of the membrane
phospholipids and this change can be closely related to the
protein arrangement followed by the steady state anisotropy of
Na/K-ATPase.
At low concentration, on various tissues, Mgo
stimulates the Na/K-ATPase 10,19,40,41; this
stimulation probably reflects the magnesium requirement for pump
phosphorylation. An excess of Mg2+ 10,40
inhibits the Na/K-ATPase, probably because of the stabilization
of E2 forms of the enzyme which reduce the rate of turnover of
the pump.
In 1991, Robinson & Pratap 42 described the
modes of inhibition of Na/K-ATPase by Mg2+. These
authors distinguish four steps at which Mg2, as a
product of the ATPase reaction, might interact as an inhibitor.
These are as follows: (1) Mg2+ bound with the
phosphorylated enzyme could be released immediately after
dephosphorylation. Mg2+ should be an uncompetitive
inhibitor of K+. With Mg2+ release
immediately preceding MgATP binding, Mg2+must be a
competitive inhibitor of MgATP. (2) An alternative, additional
step for Mg2+ release and for Mg2+ binding
as an inhibitor would be that which follows MgATP binding to (K,
Mg)E2. (3) The third possibility is that Mg2+
dissociates from (K,Mg)E1 MgATP, and binds back as a product
inhibitor. (4) The fourth possibility envisages Mg2+
release following K+ release from (K,Mg)E1 MgATP. In
this way, Mg2+ might inhibit by competing directly
with Na+ or K+ for their specific sites on
the cytoplasmic surface of the enzyme. Thus Mg2+ could
act by occupying the Na+ activating sites of the
ATPase reactions, the K+ activating sites of the
phosphatase reaction, and the monovalent cation sites that
accelerate deocclusion.
Na-K-Cl cotransport
The Na-K-Cl cotransport system moves
sodium, potassium and chloride ions across the cell membrane.
Movement is electroneutral: the number of chloride ions moved
equals the sum of the number of sodium and potassium ions. In
most cells, the ratio of ions moved is 1Na:1K:2Cl. This system is
activated by cell shrinkage and is important in regulating cell
volume and potassium content 43. In principle, the
transporter can move ions in both directions across the cell
membrane but there are some exceptions (for example, in human
amniotic membrane the cotransporter is on the fetal side only,
and in most red cells, thermodynamic constraints dictate that the
direction of net transport is inwards 45).
Ellory et al 46 indicate that the influx and efflux
of sodium and potassium across the human red cell membrane by the
bumetanide-sensitive route are inhibited by increasing
concentrations of Mg. This inhibitory effect of magnesium was not
due to a small reduction in zeta potential since the much larger
reduction in zeta potential produced by neuraminidase did not
affect transport. In the human red cell, there is a high
Mgi sensitivity of cotransport: depletion of
Mgifree inhibited and an elevation of
Mgi free increased the cotransport rate
47. In ferret red cells 48, an increase in
internal or external concentrations of magnesium stimulates
bumetanide-sensitive transport, but the system is about five
times more sensitive to internal magnesium. In the human amniotic
membrane, Mgo increases the Na-K-2CI
cotransport 19 (Fig. 5). The finding that external Mg
stimulates bumetanide-sensitive potassium uptake may indicate a
specific interaction between Mgo and the transport
system, possibly by means of a specific magnesium binding
site.
K-Cl cotransport
K-Cl cotransport is the electroneutral movement of one
K+ ion with one Cl- ion across the cell
membrane 43. K-Cl cotransport is usually measured as
the flux of K+ which depends on the presence of
Cl-, but is resistant to inhibition by ouabain and
bumetanide.
Removal of intracellular magnesium by inclusion of EDTA in the
medium markedly stimulates K-Cl cotransport whereas increasing
the concentration of Mgi inhibits it
49-51.
These experiments show that physiological changes in
intracellular ionized magnesium concentration change the activity
of K-Cl cotransport.
Na-H antiport
The plasma membranes of a wide variety of animal cells contain
a carrier-mediated transport system that brings about the
transmembrane exchange of Na+ for H+.
Recent evidence suggests that the plasma Na-H
exchanger plays a critical role in the regulation of
intracellular pH, the regulation of cell volume, the
transepithelial transport of Na+ and HCO3-,
the initiation of cell growth and proliferation in response to
activating stimuli and growth factors, and the metabolic
responses to hormones 52.
The studies by Parker et al. 53 indicate
that depleting dog red cells of Mgi prevents
activation of Na-H exchange by cell shrinkage but has
no effect on activation by acidification of the cytosol.
Replacing cell magnesium restores the activation of
Na-H exchange by cell shrinkage. The action of
magnesium would appear to be exerted either on a hypothetical
volume sensor or on the mechanism by which changes lead to
modifications in transport. On the other hand. the increase in
Mgo facilitates the Na-H exchange across
the human amnion 19. A question arises as to whether
magnesium is the transducer for cell volume changes.
By analogy, an Mg2+-H+ exchanger,
located in the apical membrane of the epithelium in the distal
colon and rumen, is fully consistent with the stimulatory effects
of volatile fatty acids on magnesium absorption at these sites
54, 55.
H-K pump
Mg2+ in the cells of periosteum and endosteum
activates the H+/K+ pump which is necessary
to keep the average pH of bone extracellular fluid slightly
higher than that of the other extracellular compartments in order
to stabilize bone 56.
Na-Ca antiporter
The Na+-Ca2+ antiporter is a membrane
carrier in the plasma membrane of certain mammalian cells that
translocates Na+ and Ca2+ in opposite
directions. The antiporter is electrogenic and the
Na:Ca2+ stoichiometry is 3:1 57.
Mg2+ was known to be a weak inhibitor
(K1=5-8 mM) of the Na+-Ca2+
antiporter in cultured heart cells 58 and cardiac
membrane vesicles 59. Moreover, Mg2+
inhibits the Na+-Ca2+ antiporter in smooth
muscle 60. These studies indicate that the inhibition
of antiporter activity by Mg2+ is due to competition
between Mg2+ and Ca2+. From this
observation and the fact that the potency depended on the
closeness of the crystal ionic radius to that of Ca2+,
it seems that the divalent cations interact with the divalent
site of the antiporter called the A site. Two classes of cation
binding sites may be distinguished: a divalent A site that binds
a single Ca2+ ion or one or two Na+ ions;
and a monovalent B site that binds the third Na+ ion
involved in the antiporter. Because the inhibition by
Mg2+ is competitive with Ca2+, it would
seem that Mg2+ binds to the A site 61.
Na-Mg exchanger
The Na+/Mg2+ system transport is not an
Na+ pathway regulated by Mg2+. but a
countertransport which permits the entry of Na+ to the
cell and the extrusion of Mg2+. A major problem with
this model is that it has been very difficult to identify the
Na+ fluxes which must accompany Mg2+
movements. In human red cell, Feray & Garay 62 may
have identified the Na+ flux by using imipramine,
which not only blocks Mg2+ but also blocks a component
of Na+ influx. Cells were treated with ouabain and
bumetanide to prevent changes in the activities of the sodium
pump or Na-K-Cl cotransport system (the major part of
the sodium transport systems in these cells), obscuring
Na+ influx through the antiport. These data suggest
that three Na+ ions move into the cell in exchange for
every Mg2+ ion. Experiments following simultaneous
changes in Na+ and Mg2+ content indicate a
stoichiometry of 2Na:lMg2+ in chicken red cells
63 and 1Na+: 1Mg2+ in rat red
cells 64 and ferret red cells 65. In frog
skeletal muscle, an Na+/Mg2+ exchange
mechanism may be involved in maintaining low levels of internal
Mg2+ concentration 66. In rat
cardiomyocytes 67, there is an
Na+-dependent amiloride-sensitive efflux. The
transmembrane gradients for Mg2+ and Na+
are coupled and an Na+/Mg2+ exchange
mechanism might be involved in the regulation of
[Mg]i, using the energy stored in the transmembrane
sodium gradient to remove Mg2+ ions against their
electrochemical gradient from the cells in exchange for
Na+ ions.
Conclusion
Magnesium may well be an important physiological regulator of
transport systems, and defects in Mg2+ metabolism may
be responsible for some modifications in transport and
cotransport systems. Intracellular Mg2+ plays an
essential role in protein synthesis and it is a key cofactor for
hundreds of enzymes. Moreover, it also modulates numerous direct,
co- and countertransport mechanisms. The interactions between
Mg2+ and Na+,K+ pathways
indicate that internal Mg2+ inhibits or blocks ionic
channels and antiport and activates or inhibits Na/K-ATPase as a
function of the concentration. Conversely, external
Mg2+ generally has an activator effect (Fig. 6).
Intracellular and extracellular Mg2+ appears to be an
essential factor in the regulation of the transport of
Na+ and K+ through the cell membranes.
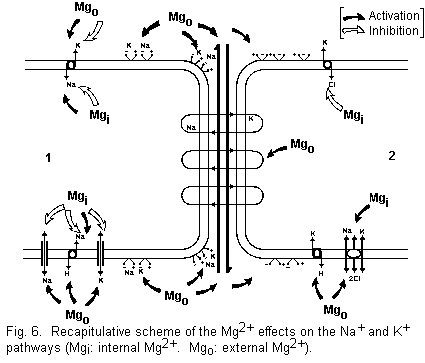
References
1. Bara, M. (1976): Magnésium et membranes.
Année Biologique 16,
290-315.
2. Nowak, L., Bregetovski, P., Ascher, P., Herbert, A. &
Prochiantz, A. (1984): Magnesium gates glutamate-activated
channels in mouse central neurones. Nature
307, 462-464.
3. Hamill, O.P., Marty, A., Neher, E., Sakmann, B. &
Sigworth, F.H. (1981): Improved patch-clamp techniques for
high-resolution current recording from the cells and cell-free
membranes patches. Pflügers Arch.
391, 85-100.
4. Mitsuiye, T. & Noma, A. (1987): A new oil-gap method
for internal perfusion and voltage clamp of single cardiac cells.
Pflügers Arch. 410, 7-14.
5. Guiet-Bara, A. & Bara, M. (1984): Cellular and shunt
conductance of human isolated amnion. I. Effect of ouabain, DNP.
amiloride and fructose. Bioelectr. Bioenerg.
13, 39-47.
6. Walser, M. (1972): Components of sodium and chloride flux
across toad bladder. Biophys. J. 12,
351-368.
7. Siegel, B. & Civan, M.M. (1976): Aldosterone and
insulin effects on driving force of Na+ pump in toad
bladder. Am J. Physiol. 230,
1603-1608.
8. Shanes, A.M. (1958): Electrochemical aspects physiological
and pharmacological action in excitable cells. Pharmacol.
Rev. 10, 59-165.
9. Aguilera, A.J., Kirk. K.L. & DiBona, G.F. (1978):
Effect of magnesium on sodium transport in toad urinary bladder.
Am. J. Physiol. 234, F192-F198.
10. DiBona, G.F. (1980): Effect of magnesium on sodium
transport in the toad bladder. In: Magnesium in health and
disease, eds. M. Cantin & M.S. Seelig, pp. 129-135. New
York: Spectrum.
11. Bara, M., Guiet-Bara, A. & Durlach, J. (1988):
Modification of the human amniotic membrane stability after
addition of magnesium salts. Magnesium Res.
1, 23-28.
12. Bara, M., Guiet-Bara, A. & Durlach, J. (1988):
Analysis of magnesium membraneous effects: binding and screening.
Magnesium Res. 1, 29-33.
13. Bara, M., Guiet-Bara, A. & Durlach, J. (1989): A
qualitative theory of the screening-binding effects of magnesium
salts on epithelial cell membrane: a new hypothesis.
Magnesium Res. 2, 243-247.
14. Pusch, M. (1990): Open-channel block of Na+
channels by intracellular Mg2+. Eur. Biophys.
J. 18, 317-326.
15. Pusch, M., Conti, F. & Stühmer, W. (1989):
Intracellular magnesium blocks sodium outwards currents in a
voltage- and dose-dependent manner. Biophys. J.
55, 1267-1271.
16. Lin, F., Conti, F. & Moran, O. (1991): Competitive
blockage of the sodium channel by intracellular magnesium ions in
central mammalian neurones. Eur. Biophys. J.
19, 109-118.
17. Hille, B. (1972): The permeability of sodium channel to
metal cations in myelinated nerves. J. Gen. Physiol.
51, 199-219.
18. Cukierman, S. & Krueger, B.K. (1990): Modulation of
sodium channel gating by external divalent cations: differential
effects on opening and closing rates. Pflügers
Arch. 416, 360-367.
19. Guiet-Bara, A., Bara, M. & Durlach. J. (1990):
Comparative study of the effects of magnesium and taurine on
electrical parameters of natural and artificial membranes. VII.
Effects on cellular and paracellular ionic transfer through
isolated human amnion. Magnesium Res.
3, 249-254.
20. Horie, M. & lrisawa, H. (1987): Rectification of
muscarinic K+ current by magnesium ion in guinea pig
atrial cells. Am J. Physiol. 253,
H210-H214.
21. Horie, M. & Irisawa, H. (1989): Dual effects of
intracellular magnesium on muscarinic potassium channel current
in single guinea-pig atrial cells. J. Physiol. (Lond.)
408, 313-332.
22. Hille, B. (1984): Mechanism of block in ionic channels
of exicitable membrane. Sunderland, MA, USA: Sinauer
Associates.
23. Matsuda, H., Saigusa, A. & Irisawa, H. (1987): Ohmic
conductance through the inwardly rectifying K channel and
blocking by internal Mg2+. Nature
325, 156-159.
24. Vandenberg, C.A. (1987): Inward rectification of a
potassium channel in cardiac ventricular cells depends on
internal magnesium ions. Proc. Natl. Acad. Sci. USA
84, 2560-2564.
25. Matsuda, H. (1988): Open-state substructure of inwardly
rectifying potassium channels revealed by magnesium block in
guinea-pig heart cells. J. Physiol. (Lond)
397, 237-258.
26. Ishihara, K., Mitsuiye, T., Noma, A. & Takano, M.
(1980): The Mg2+ block and intrinsic gating underlying
inward rectification of the K+ current in guinea-pig
myocytes. J. Physiol. (Lond) 419,
297-320.
27. Silver, M.R. & DeCoursey, T.E. (1990): Intrinsic
gating of inward rectifier in bovine pulmonary artery endothelial
cells in the presence or absence of internal Mg2+.
J. Gen. Physiol. 96, 109-133.
28. Matsuda, H. (1991): Effects of external and internal
K+ ions on magnesium block of inwardly rectifying
K+ channels in guinea-pig heart cells. J. Physiol.
(Lond) 425, 83-99.
29. Noma, A. (1983): ATP-regulated K channel in cardiac
muscle. Nature 305, 147-148.
30. Noma, A. & Shibasaki, T. (1985): Membrane current
through adenosine-triphosphate regulated K channels in guinea-pig
ventricular cells. J Physiol. (Lond.)
363, 463-480.
31. Ashcroft. F.M. (1988): Adenosine-5'-triphosphate sensitive
K channels. Annu. Rev. Neurosci. 11,
97-118.
32. Findlay, I. (1987): The effects of magnesium upon
adenosine-triphosphate-sensitive potassium channels in a rat
insulin-secreting cell line. J. Physiol. (Lond.)
391, 611-629.
33. Ashcroft, F.M. & Kakei, M. (1989): ATP-sensitive
K+ channels in rat pancreatic β-cells: modulation
by ATP and Mg2+ ions J. Physiol. (Lond.)
416, 349-367.
34. Horie, M., Irisawa, H. & Noma, A. (1987):
Voltage-dependent magnesium block of
adenosine-triphosphate-sensitive potassium channel in guinea-pig
ventricular cells. J. Physiol. (Lond.)
387, 251-272.
35. Ferguson, W.B. (1991): Competitive Mg2+ block
of a large-conductance, Ca2+-activated K+
channel in rat skeletal muscle. J. Gen. Physiol.
98, 163-181.
36. Bara, M. & Guiet-Bara, A. (1984): Potassium, magnesium
and membranes. Magnesium 3,
212-225.
37. Amler, E., Svoboda, P., Teisinger, J. & Zborowski, J.
(1988): The role of carboxyl groups of Na/K-ATPase in the
interaction with divalent cations. Biochim. Biophys.
Acta 945, 367-370.
38. Gupte, S.S., Lana, L.K., Johnson, J.D., Wallick, E.T.
& Schwartz, A. (1979): The interaction of divalent cations
with (Na,K)-ATPase J. Biol. Chem. 254,
5099-5103.
39. Amler, E., Teisinger. J. & Svoboda, P. (1987):
Mg2+-induced changes of lipid order and conformation
of (Na,K)-ATPase. Biochim. Biophys. Acta
905, 376-382.
40. Flatman, P.W., & Lew, V.L. (1983): Magnesium
dependence of sodium pump mediated transport in intact human red
cells. Curr. Topics Membr. Transp. 19,
653-657.
41. Romanini, C. Tranquilli, A.L., Valensise, H., Cester. N.,
Benedetti, G., Cugini, A.M. & Mazzanti, L. (1991): In
vitro effect of magnesium ion on Na/K-ATPase isolated from
human placenta. Magnesium Res. 4,
41-43.
42. Robinson, J.D. & Pratap, P.R. (1991):
Na+/K+-ATPase: modes of inhibition by
Mg2+. Biochim. Biophys. Acta
1061, 267-278.
43. Lauf, P.K. (1986): Chloride-dependent cation cotransport
and cellular differentiation: a comparative approach. Curr.
Topics Mernbr. Transp. 27, 87-125.
44 Bara, M. & Guiet-Bara, A. (1990): Evidence of
Na-H anuport and of a Na-K-Cl
cotransport in the membrane of the human amniotic epithelial
cells. Bioelectr. Bioenerg. 24,
361-363.
45. Flatman, P.W. (1989): Magnesium and ion transport in red
cells. In: Magnesium in health and disease, eds. Y.
Itokawa & J. Durlach, pp. 43-51. London: John Libbey.
46. Ellory. J.C., Flatman, P.W. & Stewart, G.W. (1983):
Inhibition of human red cell sodium and potassium transport by
divalent cations. J. Physiol. (Lond.)
340, 1-17.
47. Mairbäul, H. & Hoffman, J.F. (1992): Internal
magnesium, 2,3-diphosphoglycerate, and the regulation of the
steady-state volume of human red blood cells by the Na/K/2Cl
cotransport system. J. Gen. Physiol.
99, 721-746.
48. Lauf, P.K. (1985): The effects of magnesium on potassium
transport in ferret red cells. J. Physiol. (Lond)
397, 471-487.
49. Lauf, P.K. (1985): Passive K-Cl fluxes in low-K sheep
erythrocytes: modulation by A23187 and bivalent cations. Am.
J. Physiol. 249, C271-C278.
50. Brugnara, C. & Tosteson, D.C. (1987): Cell volume, K
transport and cell density in human erythrocytes. Am. J.
Physiol. 252, C269-C276.
51. Delpire, E. & Lauf, P.K. (1991): Magnesium and ATP
dependence of K-Cl cotransport in low K+ sheep red
blood cells. J. Physiol. (Lond.) 441,
219-231.
52. Aronson, P.S. (1985): Kinetic properties of the plasma
membrane Na+-H+ exchanger. Annu. Rev.
Physiol. 47, 545-560.
53. Parker, J.C.. Gitelman. H.J. & McManus, T.J. (1989):
Role of Mg in the activation of Na-H exchange in dog
red cells. Am. J. Physiol. 257,
C1038-C1041.
54. Scharrer, E. & Lutz, T. (1992): Relationship between
volatile fatty acids and magnesium absorption in mono- and
polygastric species. Magnesium Res. 5,
51-58.
55. Leonhard, S., Martens, H. & Gäbel, G. (1991):
Ruminal Mg transport. A model for transepithelial Mg movement.
In: Magnesium --a relevant ion. eds. B. Lasserre &
J. Durlach, pp. 139-143. London: John Libbey.
56. Driessens, F.C.M., Steidl, L. & Ditmar, R. (1991):
Latent magnesium deficiency in osteoporotic patients.
Magnesium Res. 4, 204.
57. Reeves, J.P. & Hale, C.C. (1984): The stoichiometry of
the cardiac sodium-calcium exchange system. J. Biol.
Chem. 259, 7733-7739.
58. Wakabayashi, S. & Goshima, K. (1981): Comparison of
kinetic characteristics of Na-Ca exchange in
sarcolemma vesicles and cultured cells from chick heart.
Biochim. Biophys. Acta 645,311-317.
59. Ledvora, R.F. & Hegyvary, C. (1983): Dependence of
Na-Ca exchange and Ca-Ca exchange on monovalent
cations. Biochim. Biophys. Acta 729,
123-136.
60. Smith, J.B., Cragoe, E.J. Jr, & Smith, J. (1987):
Na/Ca antiport in cultured arterial smooth muscle cells. J.
Biol. Chem. 262, 11988-11994.
61. Slaughter, R.S., Sutko, J.L. & Reeves, J.P. (1983):
Equilibrium of calcium-calcium exchange in cardiac sarcolemmal
vesicle. J. Biol. Chem. 258,
3183-3190.
62. Feray, J.C. & Garay, R. (1988): Demonstration of a
Na+:Mg2+ exchange in human red cells by its
sensitivity to tricyclic antidepressant drugs.
Naunyn-Schmiedeberg's Arch. Pharmacol.
338, 332-337.
63. Günther, T., Vormann, J. & Fürster, R.
(1984): Regulation of intracellular magnesium by Mg2+
efflux. Biochem. Biophys. Res. Commun.
119, 124-131.
64. Ferlay, J.C. & Garay, R. (1986): An
Na+-stimulated Mg2+ transport system in
human red blood cells. Biochim. Biophys. Acta
856, 76-84.
65. Flatman, P.W. & Smith, L.M. (1991): Magnesium
transport in red cells. In: Magnesium - a relevant ion,
eds. B. Lasserre & J. Durlach. pp. 191-199, London: John
Libbey.
66. Blatter, L.A. (1990): Intracellular free magnesium in frog
skeletal muscle studied with a new type of magnesium-selective
microelectrode: interactions between magnesium and sodium in the
regulation of [Mg]1. Pflügers Arch.
416, 238-246.
67. Günther, T. & Vormann, J. (1992):
Na+-dependent Mg2+ efflux from isolated
perfused rat hearts. Magnesium Bull.
14, 126-129.
All articles by Dr. Durlach are copyrighted, and permission is
granted to Web users only to make single hard copies for personal
use. Additional reprints should be obtained from the originating
journals. Excerpts may be used by the media with attribution to
Dr. Durlach.
This page was first uploaded to The Magnesium Web Site on
April 18, 1996
http://www.mgwater.com/
