In: Metal Ions in Biological Systems, Vol. 26: Compendium
on magnesium and its role in biology, nutrition and
physiology. H. Sigel and A. Sigel eds. Marcel Dekker Inc.
New York-Basel 1990: 744 p.2, 4, 243-247
Magnesium and Its Relationship to Oncology
Jean Durlach
64, Rue de Longchamp, F-92200 Neuilly, France
Michel Bara and Andrée Guiet-Bara
Université P. et M. Curie, F-75006 Paris, France
1. INTRODUCTION
2. INTERACTION BETWEEN CARCINOGENESIS
AND MAGNESIUM STATUS
2.1 Magnesium
Disturbances by Carcinogenesis
2.2 Effect on Carcinogenesis Induced by
Disturbances of Mg Status
3. CELLULAR INTERACTION BETWWEN CANCER
AND MAGNESIUM STATUS
3.1. Uptake of
Magnesium in Normal and Tumor Cells
3.2. Plasma Membrane, Magnesium, and
Cancer
3.3. Postmembrane and Prenucleus Magnesium
and Cancer Interactions
3.4. Nucleus, Magnesium, and
Cancer
4. SYSTEMIC MECHANISMS OF THE RELATION
BETWEEN MAGNESIUM AND CANCER
4.1 Metabolic Basis
of Interaction Between Magnesium and Cancer
4.2. Magnesium, Cancer, and Immunity
4.3. Magnesium, Cancer, and Cell
Growth
5. NEW PERSONAL DATA
5.1 Magnesium and
Anticancer Agents
5.2. Magnesium and Alcohol BR
6. GENERAL CONCLUSIONS
6.1. General Scheme
of Relationship Between Magnesium Status and Cancer
6.2. Magnesium and Cancer Research
6.3. Magnesium and Cancer
Treatment
1. INTRODUCTION
Relationships between magnesium and cancer are complex: both
Mg load and Mg deficit may produce either carcinogenic or
anticarcinogenic effects. Carcinogenesis modifies the Mg status
inducing Mg distribution disturbances which may frequently
associate a tumor Mg load with Mg depletion in nonneoplastic
tissues1-3.
The aim of this chapter is first to review the consequences of
the cancerous state on Mg metabolism and those of the
disturbances of Mg status on carcinogenesis, to analyze the
cellular and systemic bases of these relations, and to appreciate
their theoretical and practical implications. We will then
present some new personal data on the membranous relationship
between magnesium and various anticancer or carcinogenic agents.
We will attempt as a conclusion to show how these data may bring
about new promising developments in cancer research as well as in
Mg and anticancer treatment.
2. INTERACTION BETWEEN
CARCINOGENESIS AND MAGNESIUM STATUS
2.1. Magnesium Disturbances by
Carcinogenesis
During chemical carcinogenesis it is possible to observe Mg
cellular deficit in preneoplastic and neoplastic states. Mg
deficit causes structural and functional alterations of the
plasma membrane with a decrease of the intracellular cations Mg
and K and an increase of the extracellular cations Ca and Na in
neoplastic cells. These drastic changes are grossly similar to
the cellular alterations observed in magnesium
deficiency1-4. One of the principles for the
prophylactic use of Mg in oncology is to avoid the facilitation
of the effect of a carcinogenic agent by the induction of
cellular disturbances of this type1-4. In fact, the
disorders in Mg distribution in carcinogenesis are far more
complex than those observed in simple Mg deficiency. During the
neoplastic state, Mg bound in intracellular structures decreases
for the most part while its concentration in the
cytosol1-4 -- and sometimes in
mitochondria5 -- increases. This type of disturbance
in Mg distribution, unobserved in simple Mg deficiency, indicates
an alteration of the intracellular Mg regulation, i.e.,
a type of Mg depletion even at an early stage of
carcinogenesis1-3. At a later stage, disturbances in
Mg distribution are even more complex. In man, at a later
cancerous stage, the disturbance in Mg distribution associates an
increased Mg level in the tumor1-4,6-8 and in blood
cells including erythrocytes and lymphocytes1-4,6,
with some stigmata of Mg depletion which differ according to the
clinical type and treatment1-4,6-13. For example, the
Mg level of breast cyst fluids does not constitute a risk
indicator for breast cancer14. A high concordance
exists between high Mg/Ca ratio levels in meningiomas and the
absence of microscopic calcifications, which may testify to the
inhibitory potency of Mg against mineralization15. Mg
depletion appears more frequently in patients with solid tumor
malignancy than with hemopathic malignancy16.
Radiation enteritis increases fecal Mg loss3,17 and
cancer cachexia urinary Mg loss18 in particular. Mg
depletion does not occur equally in all tissues. Experimental and
clinical data show that usually the adverse effect of Mg
depletion in nonneoplastic tissue are
reversible1-3,17,19.
It seems, therefore, that an established cancer induces Mg
disturbances which cause Mg load in tumoral tissue, possibly due
to Mg mobilization through blood cells, with Mg depletion in
nonneoplastic tissue1-4,6.
2.2. Effect on
Carcinogenesis Induced by Disturbances of Mg Status
Mg deficiency may produce either tumorogenic or
anticarcinogenic effects. In reverse, Mg acts either as an
anticancer or a carcinogenic agent.
2.2.1. Tumorogenesis Due to Mg Deficit and Anticancer
Effects of Mg
In the rat only, a prolonged Mg deficiency induces the
development in a minority of animals of either benign lesions
(tumor-like connective proliferation in the
intestine1,2) or periosteal desmoid tumor
formation20, or malignant defects thymic lymphoma and
myeloid leukemia1-4, 6,11). There is no correlation
between the development of benign and malignant proliferations.
Genetic factors are prominent: Mg deficiency does not induce
oncogenicity in any species other than the rat, or in any rat
except in a minority of rats of the same particular
strains1-3,21. The anticarcinogenic properties of Mg
observed on this particular model provide only poor support to
the general notion of the anticarcinogenic properties of Mg. In
several models of benign or premalignant tumorogenesis, Mg
inhibits the development of the defect: on established benign
hyperplastic lesions induced by topical application of DMBA
(7,12-dimethylbenzanthracene) in hamster cheek pouch2
and on two premalignant esophageal tumors induced in rats either
by MBN (N-nitro-somethylbenzylamine)22 or mainly by a
corn-fed diet23 of the same type as that of the Zulu
population with a high risk of esophageal cancer24. It
is very important to emphasize that the preventive effect of Mg
exists only at the early stage of tumorogenesis. This interesting
experimental notion inspires study of the prophylactic
anticarcinogenic power of Mg, particularly in populations where a
high risk of tumorogenesis and a marginal or insufficient intake
of Mg coexist25, i.e., in a population with
colorectal polyps26. Usually, Mg supplement alone does
not favorably influence the development of established
cancer1-4,6 with the only exception being an
inhibition of the development of MAM (methylazoxymethanol
acetate)-induced large-bowel carcinogenesis in rat27
with high cathartic doses of magnesia.
2.2.2. Inhibition of Tumorogenesis Due to Mg Deficit and
Carcinogenic Effects of Mg
It has been well established for almost 25 years that Mg
deficiency antagonizes tumor implantation and inhibits growth of
induced or spontaneous tumors in the rat. In reverse, Mg intake
stimulates tumor development in the rat1-4,6,28-31.
Concurrently, Mg deficiency--associated with K
deficiency--induced by diet and hemodialysis has been used in man
as an anticancer treatment to slow the progression of inoperable
cancer. With questionable therapeutic efficiency, such attempts
carry the risk of obtaining results at the price of distressing
secondary effects1-3,6.
These data on the effect of Mg deficiency in several
neoplastic states show that Mg seems to be anticarcinogenic in
the case of early premalignant states and of some
hemolymphoreticular malignancies, and most often to be
carcinogenic in the case of solid tumors1-4,6.
3. CELLULAR INTERACTION
BETWEEN CANCER AND MAGNESIUM STATUS
The metabolism of proliferative and normal cells is markedly
different and the links between cancer and magnesium status may
rely on cellular and subcellular backgrounds.
3.1. Uptake of
Magnesium in Normal and Tumor Cells
The concentration of intracellular free Mg is mainly regulated
by adaptations of Mg efflux. When the intracellular free Mg is
Mg-depleted, nongrowing cells do not take up Mg. The uptake of
cellular Mg is possible when the normal cells are moderately
depleted and are growing. But tumor cells, e.g., Ehrlich
or Yoshida ascites tumor cells, or tumorogenic pancreatic β
cells or thymocytes rapidly reaccumulate Mg. This Mg influx in
tumor cells is inhibited sometimes by furosemide33,
sometimes by amiloride35. Newly transported
Mg2+ exchanges extensively with cytoplasmic Mg
suggesting that compartmentation of Mg2+ may be
dependent on proliferative status. Any alteration of Mg
distribution in the cancer cell may play an important role in the
neoplastic development at the membrane, cytosol, or nucleus
levels1-3,32-35.
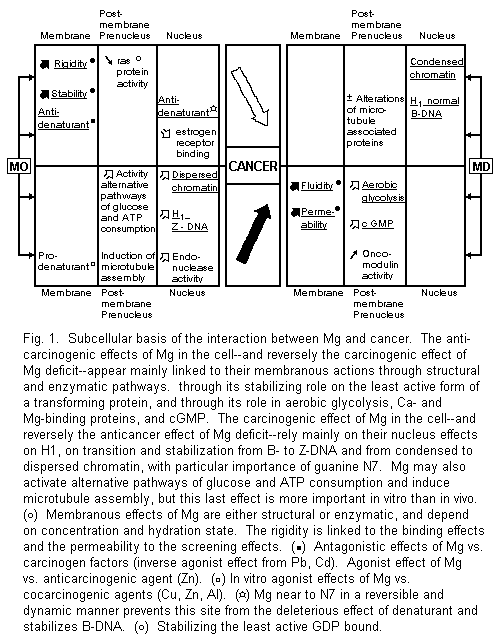
3.2. Plasma
Membrane, Magnesium, and Cancer
Accumulated evidence suggests that there is a common feature
underlying the diversity of tumors, i.e., the
alterations of their biomembrane system. It is very interesting
to point out the similarities between the plasma membrane
impairment due to malignancy and magnesium deficiency. Both
increase fluidity and permeability1-4,6,36. The
stabilizing properties of Mg are due first to its structural
role. Mg is necessary for the insertion of proteins into
membranes and for the formation of the phospholipids with
parallel electrostatic polarizing effects. The rigidifying action
of Mg is tied to the decrease of the motional freedom of
phospholipids due to their binding effect with Mg. Conformational
changes induced by Mg often depend on Mg-dependent transitions.
For example, the Mg ion exerts a central role in the regulation
of membrane receptors, promoting interconversion from monomer to
dimer forms1-3,36-38. However, this molecular basis
for membrane Mg actions does not exclude enzymatic pathways.
Neoplastic transformation of multiple cell lines reduces the
activity of numerous membrane Mg-dependent enzymes,
i.e., Na+, K+-ATPase and
Ca2+, Mg2+-ATPase 1-3, 39, 40.
All these data are relevant to the anticarcinogenic properties of
Mg.
Our recent in vitro studies on the human amniotic
membrane model showed that Mg -- at physiological dose range
-induces a biphasic effect on the maternal and fetal side. Ionic
fluxes first decrease, then increase, but without alterations of
the flux ratio. This screening effect followed by a binding
effect seems closely linked with hydrated and dehydrated forms of
Mg2+ ions41,42. A partial approach of these
observations may involve hazardous conclusions. Several
mechanisms of the anticancer action of Mg may be suggested. For
example, since a part of the screening effect of Mg2+
may provene from stimulation of gap cellular communication, it
may antagonize the inhibition of this process considered as a
promotion phase of carcinogenesis43. Since all
carcinogenic and cocarcinogenic agents induce a decrease of ionic
fluxes with alteration of the flux ratio, the reverse actions of
the binding effect of Mg2+ might act as a general
antidote of these pollutants. Our earlier and recent data have
well established the erroneous character of these hypotheses. The
action of Mg is biphasic and should not be restricted to its
binding effect. Mg may sometimes be a competitive or
noncompetitive antagonist vs. carcinogenic pollutants,
but is may also be inactive on them and sometimes even act as a
specific or nonspecific agonist1-3,45. These
discrepancies may be partly accounted for by dissimilarities in
hydrated ionic radii, but the importance of the selected models
and of the experimental conditions should not be overlooked. For
instance, various cations such as Mg2+,
Ca2+, Zn2+, and Pb2+ may either
compete with or replace each other1-3,28,44-46 which
may have anticancer or carcinogenic implications.
3.3. Postmembrane
and Prenucleus Magnesium and Cancer Interactions
These postmembranous interactions can either be
anticarcinogenic or carcinogenic.
3.3.1. Postmembrane and Prenucleus Anticarcinogenic
Actions of Mg and Carcinogenic Effects of Mg Deficit
In cancer cells, the Ca/Mg ratio-dependent aerobic glycolysis
enhancement is mediated by phosphofructokinase activation. This
is the key enzyme in the regulation of glycolysis. Ca load and Mg
deficiency might be responsible for its
activation1,2,4.
A basic effect caused by Mg deficiency is an increased cGMP
level and cGMP seems capable of stimulating normal and malignant
cell growth1-3.
Mg stabilizes the least active GDP-bound form of the
transforming protein induced by ras
genes47.
Since oncomodulin, a tumor bivalent-cation binding protein has
calmodulin-like activity, the possibility of a Mg antagonism can
also be considered there1-3, but reversely Mg may also
stimulate the effects of calmodulin and Ca on growth regulations
acting as a carcinogenic agent48.
3.3.2. Postmembrane and Prenucleus Carcinogenic Actions of
Mg and Anticarcinogenic Effects of Mg Deficit
Mg2+ is able to modify the energy metabolism of
Ehrlich ascites tumor cells and to activate alternative pathways
responsible for glucose and ATP consumption. It decreases lactate
and pyruvate production, and increases glucose consumption with a
rapid drop of ATP levels associated with an unexpected parallel
drop of the other two nucleotides, ADP and AMP49.
Mg2+ can usually replace several polyamines and
putrescine in particular in many reactions. But it is not the
case that the tumorogenic property of putrescine reestablishes
the growth rate of Ehrlich ascites tumor cells inhibited by
α-difluoromethylornithine, an enzyme-activated irreversible
inhibitor of ornithine decarboxylase50.
In several in vitro models, Mg2+ is
critical for induction of microtubule protein assembly including
tubulin and microtubule-associated proteins. It stimulated its
polymerization and then permits both elongation and nucleation
reactions, facilitating mitosis, cell division, and
growth51,52. Nevertheless, in the Mg-deficient rat its
importance in vivo seems abated, with only minor
alterations in the associated protein and without any changes in
the polymerization rate53.
In the above-mentioned biochemical reactions, Mg usually acts
as a carcinogenic agent.
3.4. Nucleus,
Magnesium, and Cancer
The most important recent advances concerning the relationship
between Mg and cancer rest on the findings concerning the
relation between Mg and the cell nucleus, which are most often
biphasic.
3.4.1. Mg, Chromatin, and Histones
The presence of increasing Mg2+ in intact cell
nuclei affects their spatial organization. With a laser flow
microfluorimeter coupled to a phase contrast analyzer, it is
possible to identify a shrinking process induced by
Mg2+ in the range 0.4-2.5 mM, which reaches a plateau
in the range 5-20 mM and is followed by a swelling process for
the highest concentration in the range 30-60 mM. It is tempting
to speculate that the shrinking-swelling phenomenon has a
molecular correspondence at the genome level. The same Mg ranges
are shown to affect the intercalation of the flurochrome acridine
orange into chromatin. The process of acridine orange
intercalation can be considered as a stimulation of the
accessibility of the genome to biological molecules. During the
shrinking phenomenon a high level of chromatin condensation is
preserved and during the swelling phenomenon chromatin is
dispersed, leading to an increased accessibility to the cell
genome and to the higher activation of cell functional processes.
Among the main cellular cations, Mg2+ is the one with
the most influence on the chromatin structure. It is known that
larger amounts of Na+ ions are needed to cause the
same effect1,2,54.
Histones, and histone H1 in particular, stabilize the
structure of chromatin against changes in ionic strength,
i.e., against high concentrations of Mg. This regulation
may fail when the concentration of Mg increases. The histones
exhibit an increased content of elongated left-handed helices of
the poly-L-proline II type. However, Mg deficiency in the rat at
an early stage alters the histone H1. But this disorder is
compensated for in the course of the experimental deficiency,
presumably by a subtle subcellular regulation due to a release of
Mg from other subcellular sites into chromatin. It seems to be
equally important to maintain normal levels of Mg and histone H1
in chromatin in order to benefit form their critical role in
higher order structures. The Mg2+ content of histone
fractions, namely, H1, of ascite hepatome 22A tumor cells is
higher than in normal liver cells in the mouse. Variations in
chromatin compactness is an early effect induced by chemical
carcinogens in vivo. Nickel, a potent carcinogenic
metal, induces its chromosomal damaging activity through a
preferential linking in magnesium-insoluble regions of
fractionated chromatin which include histone H1. These Mg
alterations may have an etiological role in higher order
structure disturbances1-3, 55-59.
Further studies of the mechanisms controlling cellular and
subcellular Mg distribution3 and histone
interactions55 will open new paths to cancer
research.
3.4.2. Mg, Cancer, and Nucleic Acids
In the nucleus, DNA constitutes one of the selective targets
of Mg. For example, a flow cytofluorographic technique has been
able to access in vivo increased DNA synthesis induced
by parenteral Mg injections in placenta cells of pregnant
women60. Numerous in vitro studies indicate
that hexahydrated Mg ion binds directly to the guanine N7 and O6
atoms and to the negative oxygens of the phosphate through the
water molecules of hydration. The Mg atom is found to be near the
N7 site in a reversible and dynamic manner. It might prevent this
site from being attacked by denaturants; this may be one reason
why a normal Mg status is preventive against
cancer1-3,61,62. Mg2+, as well as several
polyamines63, can generate the transition from
left-handed conformation of DNA (B-DNA) to its right-handed
conformation (Z-DNA). The synthetic polynucleotide poly (dG-M5dC)
shows a right-handed form in all samples when the ratio
Mg/nucleotides is > 190/1000 and a left-handed conformation
when the same ratio is < 45/100064. When an
increased concentration of Mg induces and stabilizes the
transition form B-DNA to Z-DNA, it may stimulate carcinogenesis.
Similarly, some well-known carcinogens such as
N2-acetylaminofluorene and Ni carcinogenic compounds may
stimulate carcinogenesis1-3,57-59. On the contrary,
some anticancer agents like cisplatin or SOAZ may act as N7-site
Mg-excess antagonists or modulators by stabilizing the B-DNA
geometry1-3,61,62. Although the effects of Mg on DNA
guanine are well known, its effects on other DNA targets, such
as, for example, adenine (where N3 seems linked to
carcinogenesis) or on RNA deserve
consideration1-3,62.
Thus the relationship between Mg and nucleic acids confirms
the importance of the regulation of subcellular Mg
distribution1-3, particularly on the N7 site of
guanine: when normal, it protects against carcinogenesis; with
abnormally high levels, it generates the Z conformation of DNA
which is correlated with carcinogenicity.
3.4.3. Mg, Cancer, and Other Nuclear Fractions
Several endonucleases stimulated by Mg (and Ca) seem to be
involved in cell proliferation and can be useful enzymatic probes
for detecting chromatin changes65,66. In experimental
and human mammary tumors, Mg and nuclear estrogen receptor are
linked. Mg2+ ions inhibit the nuclear binding of the
receptor67,68. These data are scarce but seem
promising for new directions of research.
4. SYSTEMIC MECHANISMS OF THE
RELATION BETWEEN MAGNESIUM AND CANCER
Mg may interfere with malignancy through its multiple roles in
all the systems of the organism but mainly through its metabolic,
immunological, and growth-stimulating effects.
4.1. Metabolic
Basis of Interaction Between Mg and Cancer
These metabolic interactions are anticarcinogenic and
carcinogenic (Fig. 2).
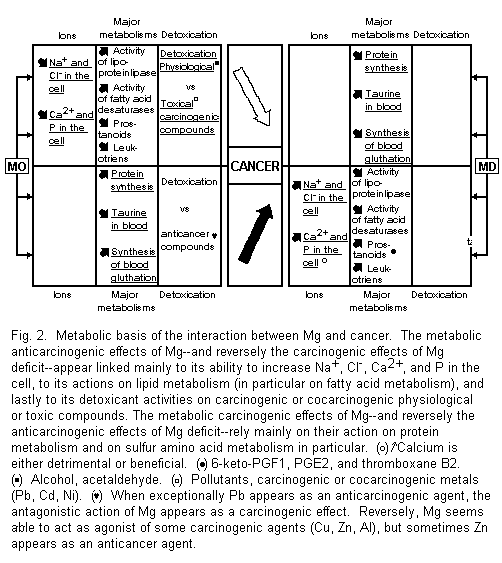
4.1.1. Metabolic Anticarcinogenic Actions of Mg and
Carcinogenic Effects of Mg Deficit
Among the drastic changes in cancer and magnesium-deficient
cells an increase of Na (and Cl) and of Ca (and P) involve
grossly similar features. Mg load may tend toward a reduction of
these ionic cellular increased concentrations1-4,38.
High levels of Na+ and Cl- are associated
with rapid cell reproduction and transformation. The amiloride
caused decrease of these ions is correlated with a decreased cell
proliferation69-72. The significance of calcifications
in tumor cells is more dubious, most often detrimental, but also
protective1-4,15,38,73.
Mg deficiency produces several variations in lipid metabolism
which may participate in carcinogenic processes, e.g.,
reduced activity of the lipoprotein-lipase system and
disturbances of fatty acid metabolism, involving inhibition of
several steps of desaturation, increased synthesis of prostanoids
(i.e., in the Mg-deficient rats significantly higher
concentrations of 6-keto-PGF-1α, PGE2, and thromboxane B2)
and leukotriene B41-3,38,74-77. It is interesting to
stress the similar striking elevation of thromboxane B2 in
thymocytes from magnesium-deficient rats, which are capable of
developing thymic lymphoma and myeloid leukemia, and from AKR
mice (8-12 months old) developing leukemia75,78.
Although magnesium deficiency in men and rat causes a
magnesium-curable hyperaggregation of platelets3,74,
hyperaggregation of platelets due to tumor cells does not appear
to be Mg-dependent79.
The metabolic anticancer properties of Mg may come from its
antagonism against carcinogenic or cocarcinogenic agents. This
antidenaturant power concerns physiological or toxic
compounds1-3,29,30,45,56-59,61,62,84,85. Both our
earlier and our recent data in vitro on human amnion in
particular and in vivo in mice have shown such
antagonistic properties against alcohol, and acetaldehyde, Cd and
Pb at different competitive sites; and Ni by noncompetitive
action1-3,45,80-82. In a few cases where Zn acts as an
anticarcinogenic agent46,86,87, its activation by
Mg45 appears as an anticancer effect.
All these metabolic actions of Mg are relevant to its
carcinogenic effects.
4.1.2. Metabolic Carcinogenic Actions of Mg and
Anticarcinogenic Effects of Mg Deficit
Among the numerous effects of Mg on protein metabolism, which
plays a prominent part in mechanisms of cell growth, its role in
sulfur amino acid metabolism deserves particular attention. The
inhibition of tumor growth due to Mg deficiency in the rat may
depend on mobilization of taurine and inhibition of the
biosynthesis of blood glutathione1-3,37, which
evidently depends on sulfur amino acid intake and
metabolism83.
Our recent in vitro observations on human amnion of
an activation of three cocarcinogenic agents on common sites for
Al and Cu, and without specificity for Zn, may indicate the
possibility of an agonistic action of Mg against some
denaturants. In exceptional cases where Pb appears as an
anticarcinogenic agent28, the antagonistic action of
Mg appears as a carcinogenic effect. These metabolic actions of
Mg agree with some carcinogenic powers of this ion.
4.2. Magnesium,
Cancer, and Immunity
Recent advances concerning the systemic interaction between Mg
and cancer rely on the close and complex links between Mg and the
immune system1-3,88-90. They are anticarcinogenic and
carcinogenic (Fig. 3).
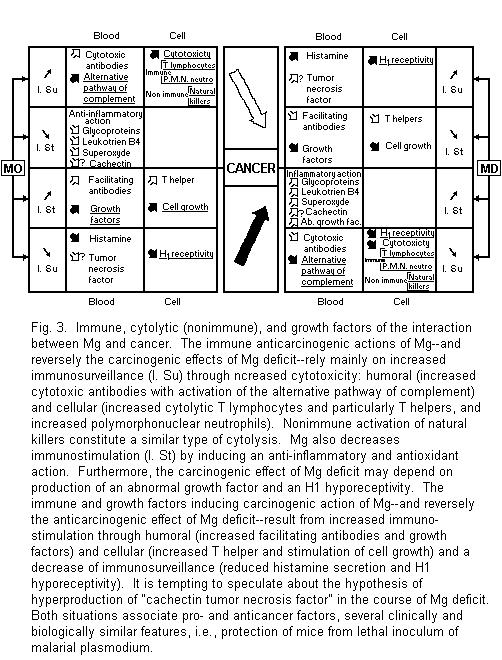
4.2.1. Immune Anticarcinogenic Action of Mg and
Carcinogenic Effect of Mg Deficit
The anticarcinogenic action of Mg rests of Mg stimulation of
cancer immunosurveillance and inhibition of immunofacilitation.
Reverse mechanisms concern the carcinogenic effects of Mg
deficit. The paths are either humoral or cellular.
Mg stimulates synthesis of cytotoxic antibodies.
Simultaneously its activation of an alternative pathway of
complement increases their inhibiting effect on tumor
growth1-3,88-90,91-93. Mg-dependent immune tumor
cytolysis is mediated by blood monocytes, mainly by T
lymphocytes, and by polymorphonuclear neutrophils with a
mechanism of tumor cell injury due to activated
oxygen1-3,88-90,94-95. Nonimmune antitumor
surveillance mediated by natural killers also requires
Mg2+ for target cell lysis1-3,84.
Study of immune disturbances of Mg deficiency shows the
reverse mechanisms of cancer immunosurveillance inhibition and
allows better identification of the type of T cytolytic
lymphocyte concerned. The percentage of total T lymphocyte and
that of T lymphocyte with helper function are reduced whereas the
percentage of T lymphocyte with suppressor function is
enhanced21. Futhermore, Mg deficit induces an
oncogenic inflammatory reaction with an increase of phlogistic
glycoprotein, leukotriene B4, and superoxide
anion3,77,88-90,96,97. Lastly, the same low
responsiveness of histamine releasers is similarly noted in
spleen cells of rat with experimental Mg deficiency and in rat
basophilic leukemia cells98. These immune data and
those concerning nonimmune natural killers are consistent with
some anticancer properties of Mg.
4.2.2. Immune Carcinogenic Actions of Mg and
Anticarcinogenic Action of Mg Deficit
Among the major features in cancer immunosurveillance and in
experimental and clinical magnesium deficiency, hyperhistaminemia
and histamine hyperreceptivity appear to be similarly important.
Part of the anticarcinogenic action of Mg deficit may be compared
with clinical improvement observed in some advanced cancer
diseases after a treatment combining histamine and H2
antihistaminics. This mechanism does not exclude a hypothetical
increased level of the tumor necrosis factor in blood nor an
inhibition of the immunofacilitation due to the immune
carcinogenic actions of Mg. Mg may stimulate synthesis of
antibodies and of T lymphocytes which induce tumor
growth1-3,88-90,98-100.
These data are relevant to some antitumor effects of Mg
deficit. It seems interesting to emphasize a number of
similarities between the effects of hyperproduction of cachectin
and those observed in a Mg-deficient state. Both situations
associate pro- and anticancer factors. Both reduce activity of
lipoprotein lipase and blood pressure, may lead to disseminated
intravascular coagulation, induce an imflammatory reaction with
an increased level of leukotrienes, similarly alter membrane
permeability, activate polymorphonuclear leukocytes, and lastly
protect mice from lethal inoculum of malarial plasmodium1-3,
101, 102. It is therefore tempting to speculate regarding
the hypothesis of a hypersecretion of "cachectin tumor necrosis
factor" during magnesium deficit.
Immunostimulation and immunosuppression may be either useful
or noxious in cancer treatment. The beneficial or detrimental
effects of Mg load and Mg deficit agree with this notion. But, in
fact, hemolymphoreticular malignancies are usually under
immunosurveillance: in such cases the Mg stimulating effects are
useful1-3,8. However, most often solid tumors are
immunostimulated. It is therefore advisable to be careful with
all types of immunostimulation and particularly with Mg
treatment. This rule admits some exceptions, e.g., the
control of a badly tolerated Mg deficit, especially when it is
induced by a side effect of an efficient cytolytic treatment like
cis-Pt 1-3.
4.3. Magnesium,
Cancer, and Cell Growth
Indirectly through mediation of growth factors and directly
through its effects on the cell, Mg stimulates normal and
proliferative cell growth. This action constitutes one of the
main mechanisms of its carcinogenic property.
4.3.1. Direct Effect of Mg on Cell Growth
Magnesium is an essential factor of cell growth. Free
Mg2+ permits tubulin polymerization and subsequent
spindle formation. It triggers chromosome condensation and
eventually spindle breakdown, enabling nuclear and cell division
to proceed. In tumor cells, uptake of Mg and cell growth are
coordinated processes. Mg plays a central role in the
proliferative response1-3,38,47,103-105. Its effects
are more deleterious because the Mg requirement in transformed
cells is less than in normal cells1-3,106.
4.3.2. Magnesium, Cancer, and Growth Factors
Growth factors in oncology may induce two noxious effects:
either through their mitotic actions by stimulating cell
proliferation or through neoangiogenesis by acting as angiogenic
factors107. Mg mediates and promotes the effects of
several serum-derived hormone-like growth factors, epidermal
growth factors, and nerve growth
factors1-3,106-110.
Unexpectedly, Mg deficiency-induced malignant T-cell lymphomas
produce a particular protein growth factor with a molecular
weight of 20'000-25'00021. These stimulating effects
of Mg on growth may participate in some of its carcinogenic
properties, but paradoxically a particular growth factor may also
intervene in some carcinogenic properties of Mg
deficiency-induced lymphoma.
5. NEW PERSONAL DATA
Previous model studies1,2,45 on the human amniotic
membrane, a leaky, asymmetric, and nonexcitable membrane,
indicated that Mg is not an antagonist of all carcinogenic
metals. These data have shown the essential role of the membrane
in the case of a competitive inhibition between Mg and metals,
but also another action level, e.g., the nucleus and
nucleic acids, in the case of a noncompetitive inhibition and an
absence of action.
The present data determine the relationship between Mg and
anticancer agents, and between Mg and alcohol.
5.1. Magnesium and
Anticancer Agents
5.1.1. Magnesium and Anticancer Metals or
Metalloids
In recent studies1,2 the relationship between Mg
and two anticancer metals, i.e., gallium (GA) and
cisplatin (Cis-Pt), has been observed. Ga and Cis-P5 decrease the
ionic permeability through the human amnion, estimated by the
measurement of the total ionic conductance (Gt) on the two sides
(maternal and fetal), except that Cis-Pt already does this at low
concentration on the fetal side. There is a noncompetitive
inhibition between Mg and Ga, i.e., Mg and Ga are fixed
on different sites on the surface membrane. Moreover, there is no
antagonism between Mg and Cis-Pt.
Selenium, a metalloid, may be considered with regard to its
anticancer properties. The Se-mediated anticarcinogenesis seems
to be due to an action on glutathione, and this action is
identical to the effects observed in the case of magnesium
deficiency1. In the human amnion, Se decreases Gt on
the maternal side, but has no effect on the fetal side (Fig. 4).
There is a noncompetitive antagonism between Mg and Se on the
maternal side (Fig. 5); Mg and Se are fixed on different sites on
the surface membrane.
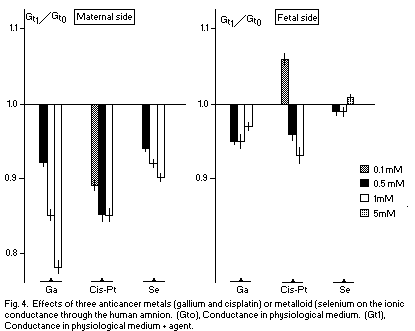
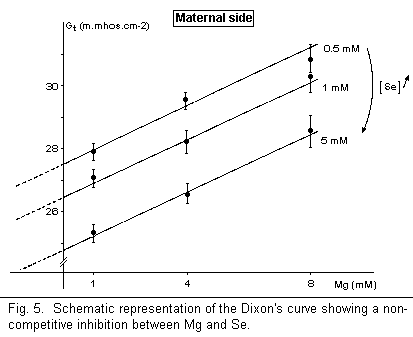
The reduction of the membrane conductance Gt, a deleterious
effect, observed in these data indicates that the
anticarcinogenic effects of Ga, Cis-Pt, and Se are located on
targets other than the plasmatic membrane. The noncompetitive
antagonism (Mg-Ga, Mg-Se) and the absence of action (Mg-Cis-Pt)
may indicate that the sites of anticancer agents and Mg are
localized in another part of the cell (Cis-Pt may control the Mg
level on N7 of guanine in DNA, for example) if their antitumoral
effects are due to competitive actions of Mg.
5.1.2. Magnesium and Anticancer Antibiotic
Mithramycin is an antitumor antibiotic3 that can
basically be considered to have powerful properties of the same
type as calcitonin. Mithramycin can in fact cause a rapid and
severe lowering of the blood magnesium level.
In the human amnion, mithramycin reduces Gt on the maternal
side but has no effect on the fetal side. There is no antagonism
between Mg and mithramycin. These results limit the role of the
membrane in the effects of mithramycin.
5.2. Magnesium and
Alcohol
Alcohol may be classified among the carcinogenic or
cocarcinogenic factors. Alcohol in vitro, like
carcinogenic metals, decreased Gt2. There is a
preventive antagonism between Mg and alcohol on the maternal
side, and a curative opposing action, independent from Mg
concentration, on the fetal side and regardless of Mg
concentration on the maternal side81,82.
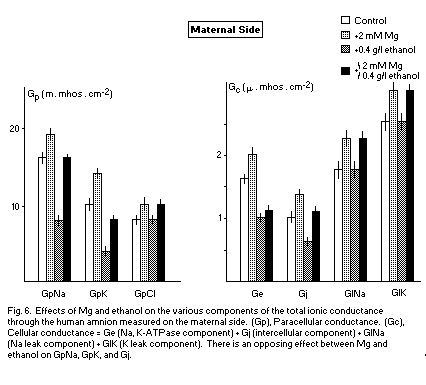
The transamniotic conductance Gt is the sum of various
components: paracellular (GpNa, GpK, GpCl) and cellular
(Na,K-ATPase component; Ge, intercellular component; Gj, leak
component G1Na, G1K).
These various components are reduced by alcohol and increased
by Mg. This opposite action may indicate an antagonism between Mg
and alcohol on all components of Gt. At a concentration of 2mM
(corresponding to a binding state41,42) (Fig. 6) in
preventive and opposing action, Mg antagonizes the deleterious
effects of alcohol on GpNa, GpK, and Gj on the maternal side. In
this case, the membranous effects are important and Mg may be
considered as an anticarcinogenic factor.
6. GENERAL CONCLUSIONS
The data described may provide a basis for further
developments in the physiological, experimental, and therapeutic
fields.
6.1. General Scheme
of Relationship Between Magnesium Status and Cancer
Through numerous cellular and systemic mechanisms, the
relation between Mg status and cancer appears very complex: both
Mg load and Mg deficit sometimes induce anticancer and sometimes
carcinogenic effects (Fig. 1-3).
The following general scheme may be retained. Established
carcinogenesis induces Mg disturbances which associate tumor Mg
load due to Mg mobilization through blood cells with Mg depletion
in nonneoplastic tissue. At a premalignant stage and in the case
of some hemolymphoreticular malignancies, Mg deficiency seems
carcinogenic, but in the case of solid tumors it inhibits tumor
development1-3. These various disturbances entail
several logical consequences concerning cancer research and
treatment.
6.2. Magnesium and
Cancer Research
In experimental and clinical cancer and anticancer research,
it appears very important to investigate magnesium status and the
homeostatic of perturbating factors which regulate or disturb
cellular, subcellular, and systemic Mg metabolism. We still have
limited knowledge of these processes1-3, but the role
of taurine provides a first approach3,37,111-115.
6.3. Magnesium and
Cancer Treatment
Indications for cancer therapy of Mg supplementation of of
induced Mg deficiency are critical. But it is possible to
speculate on the importance of ideal treatment for Mg
disturbances in an overall cancer treatment.
6.3.1. Contraindictions and Indications of Mg
Therapy
Within the context of our present knowledge, we will consider
malignant solid tumors as a relative contraindication of
magnesium therapy since their development may be stimulated by
Mg. Nevertheless, if Mg depletion induces an important
discomfort, one may use palliative Mg therapy to improve the
quality of life. This is more permissible as the risk of inducing
Mg excess in the sites where it is noxious is
prevented--e.g., in the case of efficient cytostatic
treatment--by Cis-Pt. Moreover, Mg supply may also decrease both
toxicity and anticancer efficiency of the
drug1-3,17,116-121, i.e., for cytostatic
anthracycline122,123.
Mg supply may be used as an adjuvant treatment in
hemolymphoreticular malignancies, particularly when symptomatic
Mg deficit appears, aggravated by iatrogenic factors,
i.e., use of therapies inducing Mg deficit: cyclosporine
for marrow transplantation, aminoglycosides, abdominal
irradiation, several cytostatic
drugs1-3,17,116-123.
Prophylactic anticancer action requires a balanced, normal Mg
metabolism, with sufficient Mg intake in
particular1-3.
6.3.2. Induction of Mg Deficit
The induction of severe Mg deficiency through dialysis
associated with a diet deficient in Mg, even associated with K
deficiency, is the least advisable as it is of dubious
efficiency1-3. The use of Mg antagonists that
accumulate specifically in tumor tissue would be a more appealing
alternative. Gallium would fill this bill but its tolerance and
effectiveness are still under study1-3,124,125.
6.3.3. Ideal Treatment
In the future, the ideal treatment for Mg disturbances in
cancer should control the systemic and local factors which
regulate cellular and subcellular Mg distribution in neoplastic
tissues and those which prevent Mg depletion in extramalignant
tissues.
ACKNOWLEDGMENT
We thank Dr. Jean-Pierre Mareschi for his valuable help by
providing the most recent computerized data for this literature
survey.
REFERENCES
1. J. Durlach, M. Bara, A. Guiet-Bara, and P. Collery,
Anticancer Res., 6, 1353 (1986).
2. J. Durlach, P. Rinjard, M. Bara, A. Guiet-Bara, and P.
Collery, in Magnesium: Physiologische Aspekte für die
Praxis (B. Lasserre, ed.), Panscientia Verlag,
Hedzingen-Zürich, 1987, p. 26ff.
3. J. Durlach, Magnesium in Clinical Practice, J.
Libbey, London, 1988, p. 386.
4. L. J. Anghileri, P. Collery, P. Coudoux, and J. Durlach,
Magnesium Bull., 3, 1 (1981).
5. G. Fiskum and R. S. Cockrell, Arch. Blochem.
Biophys., 240, 723 (1985).
6. P. Collery, L. J. Anghileri, P. Coudoux, and J. Durlach,
Magnesium Bull., 3, 11 (1981).
7. Z. Szmeja and H. Konczewska, H.N.O., 34, 85
(1986).
8. J. Alexandrowicz, J. Dobrowolski, and J. Lisiewicz,
Magnesium Res., 2, 83 (1989).
9. M. Abdulla, Z. Gesamte Hyg., 30,
196 (1984).
10. M. J. Laires and J. R. D. Silva, Anticancer Res.,
5, 638 (1985).
11. L. Youngman, J. Chen, S. Liu, J. Li, R. Peto, and T. C.
Campbell, Fed. Proc., 44, 1341
(1985).
12. A. A. Fomina, Vrach Delo, 0/10,
58 (1986).
13. S. E. Shehin and M. B. Zemel, FASEB J.,
2, 3321 (1988).
14. H. L. Bradlow, F. D. Skidmore, M. K. Schwartz, M.
Fleisher, and D. Schwartz, in Endocrinology of Cystic Breast
Disease (A. Angeli, H. L. Bradlow, and L. Dogliotti, eds.),
Raven Press, New York, 1983, P. 197ff.
15. W. Muller and R. Iffland, Acta Neuropathol.
(Berl.). 55, 53 (1981).
16. T. G. Frazier, M. E. Mucha, I. H. Rush, E. J. Trull, S. A.
Carlson, and J. A. O'Connor, J. Surg. Oncol.,
13, 35 (1980).
17. L. Cohen and R. Kitzes, Magnesium, 4, 16
(1985).
18. D. W. Nixon, D. H. Lawson, M. Kutner, J. Ansley, M.
Schwarz, S. Heymsfield, R. Chawla, T. H. Cartwright, and D.
Rudman, Cancer Res., 41, 2038
(1981).
19. X. Le Loet, C. Ducastelle, J. Hemet, and P. Deshayes,
Rev. Rhum. Mal. Osteoartic., 50, 141
(1983).
20. L. F. Belanger, in 1st Int. Symposium on Mg Deficit in
Human Pathology; II. Book of Communications and Discussion
(J. Durlach, ad.), S.G.E.M.V., Vittel, 1973, P. 425ff.
21. T. Günther, R. Averdunk, K. Wonigeit, and J. Vormann,
Magnesium Bull., 10, 22 (1988).
22. S. J. Van Rensburg, J. M. Hall, and D. B. DuBruyn, J.
Natl. Cancer Inst., 75, 561 (1985).
23. S. J. Van Rensburg, J. M. Hall, and P. S. Gatherole,
Nutr. Cancer, 8, 163 (1986).
24. S. J. Van Rensburg, E. S. Bradshaw, and E. F. Rose,
Br. J. Cancer, 51, 399 (1985).
25. S. J. Van Rensburg, S. Afr. Med. J.,
S21, 9 (1987).
26. G. Macquart-Moulin, E. Riboli, J. Cornee, R. Kaaks, and P.
Berthdézène, Int. J. Cancer.,
40, 17S (1987).
27. H. Mori, T. Tanaka, and T. Kuniyasu, Magnesium
Res., 2, 85 (1989).
28. R. M. Galt, P. A. McCreary, G. H. Laing, and G. M. Hass,
in Trace Substances in Environmental Health, Vol. 14 (D.
D. Hemphill, ed.), Univ. Missouri-Columbia, 1980, P. 189ff.
29. G. M. Hass, G. H. Laing, R. M. Galt, and P. A. McCreary,
Magnesium Bull., 3, 217 (1981).
30. R. M. Galt, G. H. Laing, and G. M. Hass, in Trace
Substances in Environmental Health, Vol. 16 (D. D. Hemphill,
ed.), Univ. Missouri-Columbia, 1982, p. 205ff.
31. B. J. Mills, R. D. Lindeman, and C. A. Lang, Proc.
Soc. Exp. Biol. Med., 181, 326 (1986).
32. R. D. Grubbs, C. A. Wetherill, K. Kutschke, and M. E.
Maguire, Am. J. Physiol., 248, C51
(1985).
33. T. Günther, J. Vormann, and R. Averdunk, FEBS
Lett., 197, 297 (1986).
34. V. Hoftiezer, P. 0. Berggren, and B. Hellman, Cancer
Lett., 27, 7 (1985).
35. T. Günther and J. Vormann, Magnesium Bull.,
10, 91 (1988).
36. F. W. Heaton, S. Tongyai, and Y. Rayssiguier, in
Magnesium in Health and Disease (I. Itokawa and J.
Durlach, eds.), J. Libbey, London, Paris, 1989, P. 27ff.
37. J. Durlach, M. Bara, A. Guiet-Bara, and P. Rinjard, in
Magnesium in Cellular Processes and Medicine (B. Altura,
J. Durlach, and M. S. Seelig, eds.), Karger, Basel, 1987, p.
219ff.
38. T. Günther, in Magnesium in Health and
Disease (Y. Itokawa and J. Durlach, eds.), J. Libbey,
London, 1989, p. 3ff.
39. D. L. Guernsey and I. S. Edelman, J. Biol. Chem.,
261, 11956 (1986).
40. G. Calviello, D. Bossi, and A. Cittadini, Cell. Biol.
Int. Rep., 10, 478 (1986).
41. M. Bara, A. Guiet-Bara, and J. Durlach, Magnesium
Res., 1, 23 (1988).
42. M. Bara, A. Guiet-Bara, and J. Durlach, Magnesium
Res., 1, 29 (1988)
43. J. E. Trosko and C. C. Chang, J. Am. Coll. Nutr.,
7, 428 (1988).
44. B. S. Levine and J. W. Coburn, N. Engl. J. Med.,
310, 1253 (1984).
45. M. Bara, A. Guiet-Bara, and J. Durlach, Magnesium
Res., 1, 231 (1988).
46. B. L. Vallee, Biofactors, 1, 31
(1988).
47. E. de Vendittis, R. Zahn, and O. Fasano, Eur. J.
Biochem., 161, 473 (1986).
48. R. A. Hickie, J. W. Wei, L. M. Blyth, D. Y. Wong, and D.
J. Klaasen, Can. J. Biochem. Cell. Biol.,
61, 934 (1983).
49. A. Cittadini, F. I. Wolf, G. Calviello, and D. Bossi,
Magnesium Res., 2, 126 (1989).
50. S. M. Oredsson, S. Anehus, and O. Heby, Mol. Cell.
Biochem., 64, 163 (1984).
51. A. B. Huang, C. M. Lin, and E. Hamel, Biochim.
Biophys. Acta, 832, 22 (1985).
52. S. Roychowdhury and F. Gaskin, Biochemistry,
25, 7847 (1986).
53. P. R. Trumbo, J. D. Watkins, and W. G. Martin, Fed.
Proc., 41, 362 (1982).
54. J. Widom, J. Mol. Biol., 190,
411 (1986).
55. L. K. Tkeshelashvili, N. V. Sichinava, N. A. Sapozhnikova,
and A. N. Rcheulishvili, Biofizika, 28,
582 (1983).
56. S. R. Patierno, M. Sugiyama, J. P. Basilion, and M. Costa,
Cancer Res., 45, 5737 (1985).
57. S. R. Patierno and M. Costa, Cancer Biochem.
Biophys., 9, 113 (1987).
58. S. R. Patierno, M. Sugiyama, and M. Costa, J. Biochem.
Toxicol., 2, 12 (1987).
59. K. Conway, X. W. Wang, L. S. Xu, and M. Costa,
Carcinogenesis, 8, 1115 (1987).
60. L. Ungar, Z. Kazy, F. Boldog, J. Menyhart, and P. Siklos,
in Actual Standing In EPH Gestosis (C. Goeke, ed.),
Elsevier, Amsterdam, 1985, p. 257ff.
61. T. Theophanides and M. Polissiou, Magnesium,
5, 221 (1986).
62. T. Theophanides and M. Polissiou, in Metal-Based
Anti-Tumor Drugs (M. Gielen, ed.), Freund, Tel Aviv,
1989.
63. J. E. Morgan, J. W. Blankenship, and H. R. Matthews,
Arch. Biochem. Biophys., 246, 225
(1986).
64. S. Devarajan and R. H. Shafer, Nucleic Acids
Res., 14, 5099 (1986).
65. M. G. Giles, P. S. Rennie, and N. Bruchovsky, Mol.
Cell. Endocrinol., 45, 167 (1986).
66. N. N. Khodarev, I. A. Sokolova, S. S. Aleksandrova, I. I.
Votrin, and I. V. Chupyrina, Mol. Gen. Mikrobiol. Virusol.
(USSR), 0/9, 24 (1987).
67. J. Nelson, R. Clarke, G. R. Dickson, H. W. Vandenberg, and
R. F. Murphy, J. Steroid Biochem., 25,
619 (1966).
68. W. Dietrich, M. Gorlich, and E. Heise, Eur. J. Cancer
Clin. Oncol., 22, 181 (1986).
69. I. L. Cameron and N. K. R. Smith, Scan. Electron
Microsc., 2, 463 (1980).
70. I. L. Cameron and K. E. Hunter, Cancer Res.,
43, 1074 (1983).
71. A. Romeu, L. Arola, and M. Alemany, Cancer Biochem.
Biophys., 9, 53 (1986).
72. A. Romeu, L. Arola, and M. Alemany, Oncology,
44, 319 (1987).
73. Z. Chirulescu, C. Chirilolu, A. Suciu, and R. Pirvulescu,
Med. Interne (Romania), 25, 257
(1987).
74. Y. Rayssiguier, A. Mazur, P. Cardot, and E. Gueux, in
Magnesium in Health and Disease (Y. Itokawa and J.
Durlach, eds.), J. Libbey, London, Paris, 1989, p. 27ff.
75. S. Nigam, R. Averdunk, and T. Günther,
Prostaglandins Leukotrienes Med., 23, 1
(1986).
76. F. Garaci, R. Paoletti, and M. G. Santoro,
Prostaglandins in Cancer Research, Springer Verlag,
Berlin, 1987, p. 288.
77. K. Hanada, K. Suzuki, I. Hashimoto, K. Sone, and K.
Ishida, Magnesium Res., 2, 19
(1989).
78. R. A. Karmali, T. Wustrow, H. T. Thaler, and R. A. Good,
Cancer Res., 44, 467 (1984).
79. E. Bastida, G. Escolar, A. Ordinas, S. L. Giardina, and G.
A. Jamieson, J. Lab. Clin. Med., 106,
68 (1985).
80. J. Durlach and Y. Rayssiguier, Revue Alcool.
(France), 26, 1 (1980).
81. A. Guiet-Bara, M. Bara, and J. Durlach, Magnesium
Bull., 9, 36 (1987).
82. A. Guiet-Bara, M. Bara, J. Durlach, and C. Pechery,
Alcohol, 5, 63 (1988).
83. J. Durlach, S. Poenaru, S. Rouhani, M. Bara, and A.
Guiet-Bara, in Nutrients and Brain Function (W. B. Essman, ed
Karger, Basel, 1987, p. 48ff.
84. K. S. Kasprzak, R. V. Quander, and L. A. Poirier,
Carcinogenesis, 6, 1161 (1985).
85. K. S. Kasprzak, J. M. Ward, L. A. Poirier, D. A.
Reichardt, A. C. Denn, and C. W. Reynolds,
Carcinogenesis, 8, 1005 (1987).
86. A. S. Prasad, in Zinc Metabolism (A. S. Prasad,
ed.), C. C. Thomas, Springfield, IL, 1966, p. 288ff.
87. M. Bara, A. Guiet-Bara, and J. Durlach, J. Trace Elem.
Electrolytes Health Dis., 2, 111
(1986).
88. J. Durlach, Rev. Franc. Allergol.,
15, 3 (1975).
89. F. Gaudin-Harding, Magnesium Bull.,
3, 229 (1981).
90. J. H. McCoy and M. A. Kenney, in Magnesium in Cellular
Processes and Medicine (B. M. Altura, J. Durlach, and M. S.
Seelig, eds.), Karger, Basel, 1987, p. 196ff.
91. R. A. Rudders and J. Andersen, Proc. Soc. Exp. Biol.
Med., 174, 33 (1983).
92. K. Nakanishi, B. Zbar, T. Borsos, and G. Glenn, Cancer
Res., 46, 3886 (1986).
93. E. L. Pryzdial and D. E. Isenman, Mol. Immunol.,
23, 87 (1986).
94. K. E. Muse and H. S. Koren, J. Reticuloendothel.
Soc., 32, 323 (1982).
95. A. Lichtenstein, Blood, 67, 657
(1986).
96. E. Maly, Med. Hypotheses, 11,
177 (1983).
97. M. K. Horwitt, J. Nutr., 116,
1371 (1986).
98. A. Nishio, S. Matsumoto, S. Ishiguro, and M. Noburu,
Gakujutsu Hokoku-Kagoshima Daigaku Nogakubu,
35, 189 (1965).
99. C. Burtin, C. Noirot, P. Scheinmann, L. Galoppin, D.
Sabolovic, and P. Bernard, Eur. J. Cancer Clin. Oncol.,
22, 161 (1988).
100. C. Burtin, Le Concours Médical,
111, 2 (1989).
101. B. Beutler and A. Cerami, N. Engl. J. Med.,
316, 379 (1987).
102. P. Maurois, Y. Rayssiguier, E. Gueux, and E. Deicase,
Magnesium Res., 1, 118 (1988).
103. S. M. Ribeiro and H. A. Armelin, Mol. Cell.
Biochem., 59, 173 (1984).
104. T. Günther, J. Vormann, and E. Liss, Magnesium
Bull., 7, 1 (1985).
105. M. Terasaki and H. Rubin, Proc. Natl. Acad. Sci.
USA, 82, 7324 (1985).
106. W. L. McKeehan, Fed. Proc., 43,
113 (1984).
107. R. Cariou and G. Tobelem, Presse
Médicale, 17, 2219 (1988).
108. D. R. Buck and J. Bales, J. Nutr.,
113, 2421 (1983).
109. S. Cohen, Cancer, 51, 1787
(1983).
110. T. Koike, Brain Res., 299, 293
(1983).
111. M. Hashimoto, E. Nishida, and H. Sakai, Zool.
Sci., 1, 195 (1974).
112. F. Franconi, F. Martini, I. Stendardi, R. Matucci, L.
Ziletti, and A. Giotti, Biochem. Pharmacol.,
31, 3181 (1982).
113. M. Masuda, K. Horizaka, and T. Koeda, Nippon
Yakurigaku Zasshi, 84, 283 (1984).
114. C. E. Wright, T. T. Lin, Y. Y. Lin, J. A. Sturman, and G.
E. Gaull in Taurine: Biological Actions and Clinical
Perspectives (S. S. Oja, L. Ahtee, P. Kontro, and M. K.
Paasonen, eds.), Alan R. Liss, New York, 1985, p. 137ff.
115. H. F. Pierson, J. M. Fisher, and M. Rabinovitz, J.
Natl. Cancer Inst., 75, 905 (1985).
116. N. J. Vogelzang, J. L. Torkelson, and B. J. Kennedy,
Cancer, 56, 2765 (1985).
117. D. L. Trump and L. Horvet, Cancer Treatment
Rep., 69, 259 (1985).
118. A. F. Stewart, T. Keating, and P. E. Schwartz, Am. J.
Obst. Gynecol., 153, 660 (1985).
119. J. H. M. Blom, K. H. Kurth, and T. A. W. Splinter,
Int. Urol. Nephrol., 17, 331
(1985).
120. J. C. Willox, E. J. McAllister, G. Sangster, and S. B.
Kaye, Br. J. Cancer, 54, 19 (1986).
121. D. Daugaar, S. Strandgaard, N. H. Holstein-Rathlou, P. L.
Frederiksen, U. G. Svendsen, O. Munck, and F. P. Leyssac,
Scand. J. Clin. Lab. Invest., 47, 455
(1987).
122. Z. H. Lin, S. G. Li, J. J. Ye, and Y. Zhang, in
Magnesium in Health and Disease (Y. Itokawa and J.
Durlach, eds.), J. Libbey, London, Paris, 1989, p. 53ff.
123. T. Okazaki, T. Mochizuki, M. Tashima, H. Sawada, and H.
Uchino, J. Cell. Physiol., 131, 50
(1987).
124. P. Collery, H. Millart, M. Pluot, and L. J. Anghileri,
Anticancer Res., 6, 1085 (1986).
125. R. P. Warrel, Jr., M. Issacs, N. W. Alcock, and R. S.
Bockman, Ann. Intern. Med., 107, 683
(1987).
All articles by Dr. Durlach are copyrighted, and permission is
granted to Web users only to make single hard copies for personal
use. Additional reprints should be obtained from the originating
journals. Excerpts may be used by the media with attribution to
Dr. Durlach.
This page was first uploaded to The Magnesium Web Site on
April 29, 1996
http://www.mgwater.com/
