Journal of Nutritional Medicine (1992) 3, 49-59
HYPOTHESIS
Management of Fibromyalgia: Rationale for the Use of
Magnesium and Malic Acid
GUY E. ABRAHAM MD FACN1 AND JORGE D. FLECHAS MD
MPH2
1Optimox Corporation, Torrance, CA, USA and
2 Family Practice, Hendersonville, NC, USA
Primary fibromyalgia (FM) is a common clinical
condition affecting mainly middle-aged women. Of the etiologies
previously proposed, chronic hypoxia seems the one best supported
by recent biochemical and histological findings. We postulate
that FM symptoms are predominantly caused by enhanced
gluconeogenesis with breakdown of muscle proteins, resulting from
a deficiency of oxygen and other substances needed for ATP
synthesis. We present data supporting a critical role for
magnesium and malate in ATP production under aerobic and hypoxic
conditions; and indirect evidence for magnesium and malate
deficiency in FM. After treating 15 FM patients for an average of
8 weeks with an oral dosage form with dosages of 1200-2400 mg of
malate and 300-600 mg of magnesium, the tender point index (TPI)
scores (x±SE) were 19.6± 2.1 prior to treatment and
8 ± 1.1 and 6.5 ± 0.74 respectively, after an
average of 4 and 8 weeks on the magnesium malate combination
(p<0.001). Subjective improvement of myalgia occurred within
48 h of supplementation. In six FM patients, following 8 weeks of
treatment, the mean TPI was 6.8± 0.75. After 2 weeks on
placebo tablets, the TPI values increased to a mean ± SE
of 2l.5 ± 1.4 (p.<0.001). Again, subjective worsening
of muscle pain occurs within 48 h of placebo administration. A
double-blind placebo control trial is currently
underway.
Keywords: fibromyalgia, magnesium,
malate.
INTRODUCTION
Fibromyalgia (FM) is a common clinical syndrome of generalized
musculoskeletal pain, stiffness and chronic aching, characterized
by reproducible tenderness on palpation of specific anatomical
sites, called tender points [1-3]. This condition is considered
primary when not associated with systemic causes, trauma, cancer,
thyroid diseases and pathologies of rheumatic or connective
tissues. FM is nine times more common in middle-aged women
(between the ages of 30 and 50 years) than in men [4]. FM is now
recognized as being one of the most common rheumatic complaints
with clinical prevalence of 6%-20% [4]. The association of FM
with irritable bowel syndrome, tension headache, primary
dysmenorrhea [1], mitral valve prolapse [5] and chronic fatigue
syndrome [6] has been reported.
Various treatment modalities have been tested in FM patients
with poor results: tryptophan administration worsened
musculoskeletal symptoms [7]. Ibuprofen was not better than
placebo [8]. Tricyclic agents resulted in modest improvement with
a short-lived remission in only 20% of the patients [9]. The
combined administration of ibuprofen and the anxiolytic
alprazolam to FM patients resulted in significant improvement of
disease severity and severity of tenderness on palpation [10].
However, the decrease in tenderness did not reach 50% even after
8 weeks of an open label phase following a 2 week double-blind
phase.
PROPOSED ETIOLOGIES OF FM
The century-old postulate of an inflammatory reaction in FM
[11] has not been confirmed by recent histologic examination [1].
Disturbance in stage IV sleep, resulting from
tryptophan-serotonin deficiency was suggested as a possible
causative factor in the musculoskeletal pain in FM patients [12].
Plasma-free tryptophan levels in FM patients correlated inversely
with the severity of pain. Depriving normal college students of
stage IV sleep resulted in musculoskeletal symptoms similar to
those of FM patients [13]. Tryptophan administration to FM
patients did improve sleep patterns but in fact worsened
musculoskeletal pain [7].
A multifactorial etiology, with stress being the common
pathway, has been proposed [14, 15]. Elevated catecholamines are
observed in urine of FM patients [16]. However, anxiolytic agents
are of limited therapeutic value [9, 10].
Local hypoxia was postulated to play an etiologic role in the
development and the symptoms of FM [17]. Recently published
reports of clinical, morphological and biochemical pathologies in
FM patients seem compatible with this theory of chronic
hypoxia.
Patients with FM have normal muscle blood flow under resting
conditions, but decreased blood flow under aerobic exercises
[18]. Muscle tissue oxygen pressure is low in tender muscles of
FM patients, and the total mean oxygen pressure is significantly
lower than in normal controls in subcutaneous tissue of FM
patients [19], suggesting that the hypoxic condition is not
limited to the tender muscles although the hypoxia is more severe
at tender points.
Muscle biopsies from tender points showed proteolysis of
myofibrils, glycogen deposition, swollen mitochondria with
distortion of cristae and dilatation of sarcoplasmic reticulum.
[1]. Low levels of high energy phosphates such as ATP, ADP and
phosphocreatine were observed at tender points, together with
increased AMP levels [20]. The levels of high energy phosphates
were significantly lower in tender muscles than in non-tender
muscles of FM patients and in muscles of normal controls.
Decreased serum levels of several amino acids were observed in FM
patients [21].
In hypoxic muscle tissues, there is an excess of cytosolic
reducing equivalents which inhibit glycolysis. Stimulation of
gluconeogenesis occurs, with breakdown of muscle proteins and
amino acids which are used following transamination as substrates
for ATP synthesis [22, 23]. The protein breakdown observed in
muscle biopsies [1] could be the result of increased
gluconeogenesis due in part to chronic hypoxia, which has been
demonstrated in FM patients [19]. Acute viral diseases are
associated with myolysis and myalgia similar to symptoms of FM
patients [24]. The muscle pain in FM could therefore be the
result of proteolysis of muscle tissue, due to enhanced
gluconeogenesis. The low serum aminoacids [21] in spite of
increased muscle proteolysis [1] suggest a very active
gluconeogenesis in FM patients.
A HYPOTHESIS: FM IS A RESULT OF DEFICIENCIES OF SUBSTANCES
NEEDED FOR ATP SYNTHESIS
Synthesis of proteins, fats and carbohydrates necessary for
cellular integrity, normal activity and functions is dependent on
ATP availability which supplies the energy for their synthesis
and actions [25].
The synthesis of ATP by intact respiring mitochondria requires
the presence of oxygen, magnesium, substrate, ADP and inorganic
phosphate, hereafter referred to as phosphate [24]. When all
substances are present in optimal concentrations, the integrity
of the mitochondrial membrane and the capacity of the enzymatic
system in the respiratory chain become rate limiting.
The five ingredients required for the synthesis of ATP are
listed in Table 1, together with some conditions postulated to
cause a deficiency of each of these. We will review the role of
these ingredients in ATP synthesis; present data in favor of a
deficiency of some of these ingredients in FM; and demonstrate
the pivotal role of magnesium and malate in mitochondrial
membrane integrity, mitochondrial respiration and oxidative
phosphorylation, both under aerobic and hypoxic conditions; and
present preliminary data on the clinical response of 15 FM
patients to supplementation with magnesium and malic
acid.
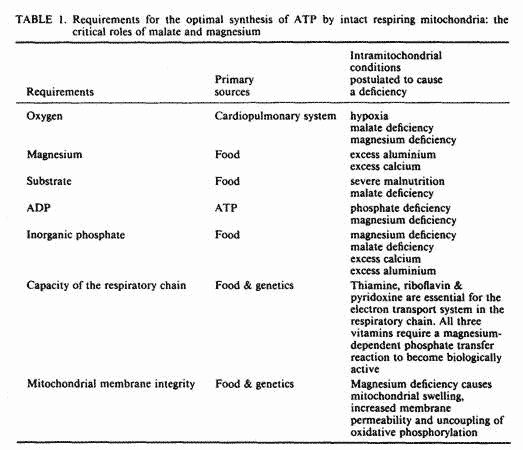
Oxygen
Anaerobic glycolysis to lactate delivers 2 moles of ATP per
mole of glucose whereas aerobic glycolysis to carbon dioxide and
water through the citric acid cycle delivers 36-38 moles of ATP
per mole of glucose [26]. Therefore, adequate oxygen supply
enhances ATP yield by 18-19 fold. The importance of oxygen for
ATP synthesis in humans has been confirmed in vivo. In patients
with chronic circulatory and/or respiratory insufficiency,
mitochondrial ATP levels were only one-half the levels found in
normal controls [27].
Relative hypoxia has been demonstrated in FM patients [19,
20]; and FM symptoms improved following aerobic conditioning
[28].
Magnesium
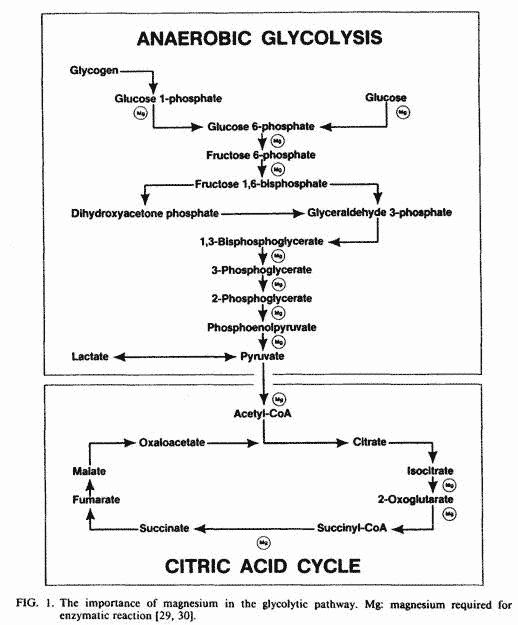
Magnesium plays a critical role in key enzymatic reactions
(Fig. 1) for both aerobic and anaerobic glycolysis [29, 30]. The
uptake and accumulation of magnesium by mitochondria is
associated with enhanced uptake of phosphate and proton extrusion
[31]. The uptake of phosphate is required for phosphorylation of
ADP, and the proton extrusion is the driving force in the
oxidative phosphorylation of ADP [26].
Through a magnesium-dependent mechanism, the mitochondria can
accumulate large amounts of CA in order to maintain low levels of
Ca in the cytosol [32]. However, this mitochondrial uptake of
calcium inhibits ATP synthesis in two ways: firstly, binding of
intramitochondrial calcium to phosphate decreases the phosphate
pool available for oxidative phosphorylation of ADP and secondly
the energy generated by the electron transport system is used up
for calcium transport, therefore, it is not available for ATP
synthesis [26]. Mitochondrial calcification eventually results in
cell death [33]. Adequate levels of magnesium are required to
maintain low cytosolic calcium [32].
Aluminium inhibits glycolysis and oxidative phosphorylation
with decreased intramitochondrial ATP and increased AMP levels
[34]. Because of its high affinity for phosphate groups,
aluminium blocks the absorption and utilization of phosphate for
ATP synthesis and, therefore may cause intramitochondrial
phosphate deficiency. Adequate magnesium levels prevent this
toxic effect of aluminium [34]. Malic acid is one of the most
potent chelators of aluminium. As an antidote to aluminium
intoxication in mice, malic acid resulted in the highest survival
ratio of several chelators tested [35]. Malic acid was the most
effective in decreasing brain aluminium levels [36].
An oxygen-sparing effect of magnesium has been demonstrated in
magnesium- deficient competitive swimmers [37]. Magnesium
supplementation lowered blood lactate levels and oxygen
consumption despite a higher glucose utilization. As will be
shown later, malate also has oxygen-sparing effect. It is
plausible, therefore that magnesium and malate deficiency could
induce a relative hypoxia in cases where the oxygen availability
is compromised, as is the case in FM patients, where blood flow
and oxygen tension are decreased.
Although magnesium status of FM patients has not yet been
reported, there is some indirect evidence in favor of magnesium
deficiency in FM patients. Magnesium deficiency causes swelling
and disruption of cristae in mitochondria, with a decreased
number of mitochondria per cell [38]. Similar mitochondrial
abnormalities have been reported in muscle biopsies of tender
points obtained from FM patients [1]. The most common symptoms
associated with FM—myalgia [39], chronic fatigue syndrome
[40], irritable bowel syndrome [41], mitral valve prolapse
[42-44], tension headache [45] and dysmenorrhea [46]— have
been reported in patients with magnesium deficiency, and
magnesium supplementation improves these symptoms.
Substrate: Pivotal Role of Malate and
Magnesium
Peripheral malate derives from food sources and from synthesis
in the citric acid cycle (Fig. 1). It plays an important role in
generating mitochondrial ATP both under aerobic [47] and hypoxic
[48, 49] conditions. Under aerobic conditions, the oxidation of
malate to oxaloacetate provides reducing equivalents to the
mitochondria by the malate-aspartate redox shuttle [47]. Under
anaerobic conditions, with an excess of cytosolic reducing
equivalents, inhibition of glycolysis occurs. By its simultaneous
reduction to succinate and oxidation to oxaloacetate, malate is
capable of removing cytosolic reducing equivalents, thereby
reversing inhibition of glycolysis [49-51]. One mole of ATP is
formed for each mole of malate reduced to succinate via fumarate
[49]. and 3 moles of ATP for each mole of malate oxidized to
oxaloacetate. Through the action of malic dehydrogenase followed
by transamination reactions, malate is converted to aspartate,
and substrates necessary for initiating transmitochondrial
exchange of metabolites through the malate-aspartate shuttle are
regenerated.
In the rat, only tissue malate is depleted following
exhaustive physical activity [52], in spite of the fact that the
other key metabolites from the citric acid cycle necessary for
ATP production remain unchanged. It has been proposed therefore
that malate deficiency is the cause of the physical exhaustion
[52], and that malate is the common mediator of increased
mitochondrial respiration by catecholamines, glucagon, and
exercise [53]. In certain bacteria which have similar
microanatomical and biochemical properties as mitochondria,
malate acts as an electron donor and generates a large proton
motive force [54], believed to be the driving force for the
mitochondrial synthesis of ATP [26].
Intraperitoneal injection of malic acid to rats in amounts of
7.5 mg per kg body weight resulted in elevated mitochondrial
malate followed by increased mitochondrial respiration, increased
mitochondrial uptake and utilization of key substrates for ATP
formation, [53]. Relatively small amounts of exogenous malate are
required to increase mitochondrial oxidative phosphorylation and
ATP production. Once an elevated mitochondrial malate
concentration is attained, it may support an increased rate of
substrate transport into the mitochondria without depleting its
own matrix concentration, for malate is regenerated in the
tricarboxylic acid cycle during the oxidation of the substrates
with which it exchanges [49, 53]. Under hypoxic conditions, there
is an increased demand for malate because malate is not only
oxidized to oxaloacetate by the action of succinate-ubiquinone
reductase [55] but also reduced to succinate [49, 51]. Increased
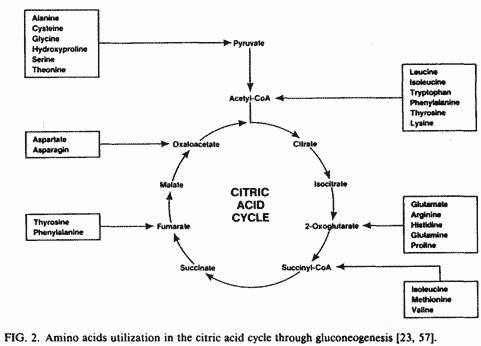
proteolysis and transamination of several amino acids occur in
order to increase mitochondrial malate levels through
gluconeogenesis (Fig. 2) [57]. Also, liver mitochondrial
phosphoenolpyruvate carboxykinase may play an important role in
generating malate from phosphoenolpyruvate and bicarbonate by a
reversal of the activity of this enzyme under conditions of
increased gluconeogenesis. This mechanism of malate production
has been demonstrated in rabbit liver [56]. The reversal of this
enzymatic reaction favors hepatic lipogenesis. Therefore, chronic
malate deficiency could play a role in certain types of
hyperlipidemia. If gluconeogenesis and possibly
phosphoenolpyruvate carboxykinase reversal cannot keep up with
malate demand, mitochondrial malate becomes deficient [52]. The
transamination of amino acids during gluconeogenesis requires
magnesium and vitamin B6. Therefore, a deficiency of these
nutrients may decrease the rate of transamination of amino acids
with a compensatory increase in proteolysis. As will be shown
later, vitamin B6 requires a magnesium-dependent phosphate
transfer reaction to become active.
In cats, under conditions of acute myocardial ischemia,
intravenous administration of sodium malate in amounts of 20-100
mg per kg body weight increases significantly coronary blood flow
without a significant increase in oxygen consumption [58]. In
rats, the oral administration of potassium malate increases
anaerobic endurance, measured by swimming time prior to
exhaustion, without a concomitant increase in carbohydrate and
oxygen utilization [52]. This effect of malate showed a
dose-response relationship with doubling of swimming time at 250
mg per kg body weight. However, at a higher dosage, a decrease in
effectiveness of malate as observed probably due to depletion of
other key substances. The above studies suggest that malate has
carbohydrate and oxygen-sparing effects.
Malate is the only metabolite of the citric acid cycle which
correlates positively with physical activity. In rats,
exercise-induced mitochondrial respiration was associated with
increased malate levels only, with the other key metabolites
remaining unchanged [53].Following endurance training of
athletes, muscles were characterized by a 50% increase in the
malate-aspartate redox shuttle enzymes [59], where malate plays a
key role. In humans as well as in other animals tested, when
there is increased demand for ATP, there is also an increased
demand and utilization of malate.
Bicarbonate loading enhanced anaerobic capacity and exercise
performance in men [60]. Although the assumed mode of action is
by an extracellular buffering mechanism, the positive effect of
bicarbonate loading could also be explained by its role as a
precursor of malate via the reverse action of phosphoenolpyruvate
carboxykinase [56]. Since gluconeogenesis favors the reversal of
phosphoenolpyruvate carboxykinase, and hypoxic conditions enhance
gluconeogenesis, bicarbonate may well serve as a precursor of
malate under hypoxic conditions.
Vitamin B6 [47] and magnesium [61] are required for normal
activity of malate dehydrogenases involved in malate-aspartate
shuttle. Phosphorylation of vitamin B6 is essential for
biological activity and this phosphate transfer reaction is
magnesium-dependent [62]. The respiratory chain involved in ATP
synthesis requires adequate amounts of the B vitamins thiamine
and riboflavin, which are the precursors of NAD and FAD
respectively [26]. These two B vitamins, like B6, require a
magnesium-dependent phosphate transfer reaction to become
biologically active. Magnesium deficiency would therefore create
a sluggish respiratory chain and a decreased efficiency in the
transfer of reducing equivalents from the cytosol to the
mitochondria.
The metabolites of the citric acid cycle and the malate
shuttle enzyme systems have not been evaluated so far in FM
patients. However, there is some evidence in favor of a
deficiency of malate dehydrogenases creating a relative malate
deficiency in FM patients. We have previously discussed the
importance of magnesium in malate dehydrogenases activity and the
evidence in favor of magnesium deficiency in FM patients.
Hypothyroidism, which is very common in FM patients, is
associated with FM like symptoms which improve following thyroid
replacement [16]. Thyroid hormones stimulate malate
dehydrogenases at the transcriptional and post-transcriptional
levels, and hypothyroidism is associated with a decrease in
malate dehydrogenases [63]. Since the transport of cytosolic
reducing equivalents in the mitochondria depends on adequate
activity of cytosolic and mitochondrial malate dehydrogenases, a
deficient enzyme system would decrease the effectiveness of
malate, creating a relative malate deficiency; this would favor
gluconeogenesis and breakdown of muscle proteins [57]. The
presence of myolysis in muscle biopsies of FM patients [1] is
highly suggestive of enhanced gluconeogenesis. In patients with
hypothyroidism and possibly in FM patients, more malate would be
required for ATP synthesis than in normal subjects. Also, it
would be expected that malate demand would be greater in
hypothyroid FM patients than in euthyroid FM patients. The use of
a metabolite of the citric acid cycle to correct an enzymatic
deficiency of the respiratory chain has proven successful in the
case of NADH coenzyme Ql0 oxidoreductase deficiency [64].
Clinical improvement was observed following succinate
administration to bypass the deficient enzymes in the respiratory
chain. A similar approach could be used to overcome a deficiency
of malate dehydrogenases by supplying bioavailable malate in
adequate amounts.
ADP
In intact living cells, ATP is usually present in a much
higher concentration than ADP and AMP. When increased workload
depletes the cell of ATP, this change in ATP/ADP accelerates
glycolysis, mitochondrial respiration and oxidative
phosphorylation of ADP to ATP. Mitochondrial ADP comes from
hydrolysis of mitochondrial ATP and ATP-mediated phosphorylation
of AMP [65]. Since these phosphate transfer reactions are
magnesium dependent, mitochondrial ADP deficiency could occur in
a metabolically active cell if magnesium and/or phosphate
concentrations were below optimal levels.
ADP deficiency has been reported in muscle biopsies of tender
points obtained from FM patients [20].
Phosphate
Phosphate uptake by the mitochondria is closely linked to the
transport of di- and tri- carboxylate anions. The net uptake of
citrate and isocitrate by the tricarboxylate transporter; and
transport of α-ketoglutarate by the dicarboxylate
transporter require an exchange with malate [66]. Therefore, the
mitochondrial uptake of phosphate depends on malate levels which
are required for exchange with phosphate. The uptake of phosphate
is also enhanced by the uptake and accumulation of magnesium by
mitochondria [31].
Intramitochondrial phosphate deficiency could occur in the
presence of low levels of magnesium and malate. Excess calcium
and aluminium could also predispose to intramitochondrial
phosphate deficiency [26, 34]. Data on mitochondrial phosphate
levels in muscle biopsies of FM patients have not yet been
published.
Integrity of Mitochondrial Membrane and Capacity of
Respiratory Chain
Magnesium plays an important role in the integrity of the
mitochondrial membrane. Magnesium deficiency is associated with
swelling of the mitochondria; increased permeability and
decreased selectivity of mitochondrial inner membrane and
uncoupling of oxidative phosphorylation [38].
As previously discussed, the respiratory chain responsible for
oxidative phosphorylation and ATP synthesis requires NAD and FAD
which derive from the vitamins thiamine and riboflavin
respectively. These B vitamins become biologically active after a
magnesium-dependent phosphate transfer reaction.
Abnormalities of mitochondrial membranes have been reported in
FM patients [1]. Adequacy of the respiratory chain, including
assessment of nutritional status with regard to the vitamins
thiamine, riboflavin and pyridoxine, have not been reported in FM
patients.
PRELIMINARY CLINICAL OBSERVATION
In an open clinical setting, 15 patients (age range 32-60)
with a diagnosis of FM based on the American College of
Rheumatology 1990 criteria [2], ingested an oral preparation
(Super Malic®, Optimox Corporation, Torrance, CA, USA),
containing 50 mg of magnesium as the hydroxide and 200 mg of
malic acid per tablet. A total daily dosage of 300-600 mg of
elemental magnesium and 1200-2400 mg of malic acid was
administered. Tender point index (TPI) [2] was assessed prior to
and following an average of 4 and 8 weeks of treatment. The mean
± SE TPI scores were 19.6 ± 2.1 prior to Super
Malic®, 8 ± 1.1 following 4 weeks; and 6.5 ±
0.74 following 8 weeks of treatment. Using paired data statistics
[67], the effect of Super Malic® on TPI values was
significant at p<0.001 at 4 and 8 weeks All patients reported
significant subjective improvement of pain within 48 h of
starting Super Malic®. Following an average of 8 weeks on
Super Malic®, six patients were switched to placebo tablets
for 2 weeks. The TPI values were 6.8 ± 0.75 and 21.5
± 1.4 prior to and following placebo administration.
Recurrence of myalgia occurs within 48 h in all the patients on
placebo tablets. This appears to be a very promising approach to
the management of FM, and a double-blind placebo-controlled trial
is currently underway.
ADDENDUM
Since submission of our manuscript, red blood cell (RBC)
magnesium levels [40] were measured in 13 newly diagnosed FM
patients [2], and the levels of RBC magnesium were below the
normal range in 12 of the 13 FM patients.
REFERENCES
[1] Yunus MB, Raman KIUP, Raman KK. Primary fibromyalgia
syndrome and myofacial pain syndrome: Clinical features and
muscle pathology. Arch Phys Med Rehabil 1988; 69: 451-4.
[2] Wolfe et al. The American College of Rheumatology 1990
criteria for the classification of fibromyalgia. Arthr Rheum
1990; 33: 160-72.
[3] Russell IJ, Vipraio GA, Morgan WW, Bowden CL. Is there a
metabolic basis for the fibrositis syndrome? Am J Med 1986; 81:
50-6.
[4] Bennett RM. Fibrositis: Evolution of an enigma. J Rheumat
1986; 13: 676-8.
[5] Pellegrino MJ, Van Fossen D, Gordon C, Ryan JM, Waylonis
GW. Prevalence of mitral valve prolapse in primary fibromyalgia:
a pilot investigation. Arch Phys Med Rehab 1989; 70: 54 1-3.
[6] Goldenberg DL, Simms RW, Geiger A, Komaroff AL. High
frequency of fibromyalgia in patients with chronic fatigue seen
in a primary care practice. Arthr Rheum 1990; 33(3) 38 1-7.
[7] Moldofsky H, Lue FA. The relationship of alpha and delta
EEG frequencies to pain and mood in “fibrositis”
patients treated with chloropromazine and L-tryptophan.
Electroencephalogr Clin Neurophysiol 1 980; 50: 71-80.
[8] Yunus MB, Masi AT, Aldag JC. Short term effects of
ibuprofen in primary fibromyalgia syndrome: A double blind,
placebo controlled trial. J Rheumat 1989; 16: 527-32.
[9] Wolfe F. The clinical syndrome of fibrositis. Am J Med
1986; 81: 7-14.
[10] Russell JI, Fletcher EM, Michalek JE, McBroom PC, Hester
0G. Treatment of primary fibrositis/fibromyalgia syndrome with
ibuprofen and alprazolam. Arthr Rheum 1991; 34: 552-60.
[11] Stockman R. The causes, pathology and treatment of
chronic rheumatism. Edin Med J 1904; 15: 107-16.
[12] Moldofsky H, Warsh JJ. Plasma tryptophan and
musculoskeletal pain in non-articular rheumatism
(“fibrositis syndrome”). Pain 1978; 5: 65-71.
[13] Moldofsky H, Scarisbrick P. Induction of neurasthenic
musculoskeletal pain syndrome by selective sleep stage
deprivation. Psychosom Med 1976; 38: 35-44.
[14] Bennett RM. Fibrositis: evolution of an enigma. J Rheum
1986; 13: 676-78.
[15) Daily PA, Biship GD, Russell JI, Fletcher EM.
Psychological stress and the fibrositis/fibromyalgia syndrome. J
Rheum 1990; 1380-85.
[16] Russell JI. Neurohormonal aspects of fibromyalgia
syndrome. Rheum Dis Clin N Am 1989; 15: 149-68.
[17] Fassbender HG, Wegner K. Morphologie und pathgenee des
weichteilrheumatismus. Z Rheuma-forschg 1973; 32: 355.
[18] Bennett RM, Clark SR, Goldberg L, Nelson D, Bonafede PR,
Porter J, Specht D. Aerobic fitness in patients with fibrositis.
Arthr Rheum 1989; 4: 454-60.
[19] Lund N, Bengtsson A, Thorborg P. Muscle tissue oxygen
pressure in primary fibromyalgia. Scand J Rheumat 1986; 15:
165-73.
[20] Bengtsson A, Henriksson KG, Larrson J. Reduced
high-energy phosphate levels in the painful muscles of patients
with primary fibromyalgia. Arth Rheum 1986; 20: 817-21.
[21] Russell JI, Michalek JE, Vipraio GA, Fletcher EM, Wall K.
Serum amino acids in fibrositis/fibro myalgia syndrome. J Rheumat
1989; 16: 158-63.
[22] Wiesner RJ, Rosen P, Grieshaber MK. Pathways of succinate
formation and their contribution to improvement of cardiac
function in the hypoxic rat heart. Biochem Med Metab Biol 1988;
40: 19-34.
[23] Rodwell VW. Catabolism of the carbon skeletons of amino
acids. In: Murray RK ed. Harper’s Biochemistry. Appleton
and Lange, 1990; 284-306.
[24] Baracos V, Rodemann HP, Dinarello CA. Stimulation of
muscle protein degradation by leukocytic pyrogen (interleukin-1),
N Eng J Med 1983; 308: 553-8.
[25] Lehninger AL. Biochemistry. Worth Publishing, 1978;
409.
[26] Lehninger AL. Biochemistry. Worth Publishing, 1978;
509-42.
[27] Bergstrom J, Bostrom H, Furst P, Hultman E, Vinnars E.
Preliminary studies of energy-rich phosphagens in muscle from
severely ill patients. Crit Care Med 1976; 4: 197-204.
[28] McCain GA. Role of physical fitness training in the
fibrositis/fibromyalgia syndrome. Am J Med 1986; 81: 73-7.
[29] Garfinkel L, Garfinkel D. Magnesium regulation of the
glycolytic pathway and the enzymes involved. Magnesium 1985; 4:
60-72.
[30] Heaton FW. Role of Magnesium in Enzyme Systems. In:
Siegel H, ed. Metal Ions In Biologic Systems. New York: Marcel
Dekker, 1990: 119.
[31] Brierly GP, Jung DW, Altschuld RA. Magnesium and
mitochondrial ion transport. In: Altura BM, Durlach J, Seelig MS,
eds Magnesium In Cellular Processes and Medicine. Basel: Karger,
1987; 89-105.
[32] Abraham GE. The calcium controversy. J Appl Nut 1982; 34:
69.
[33] Siesjo BK. Calcium and cell death. Magnesium 1989; 8:
223-37.
[34] Allen VG. Influence of aluminum on magnesium metabolism.
In: Altura BM, Durlach J, Seelig MS, eds Magnesium In Cellular
Processes and Medicine. Basel: Karger, 1987: 50-66.
[35] Domingo JL, Gomez JM, Llobet JM, Corbella J. Comparative
effects of several chelating agents on the toxicity, distribution
and excretion of aluminum. Hum Toxicol 1988; 7: 259-62.
[36] Domingo JL, Gomez M, Llobet JM. Citric, malic and
succinic acids as possible alternatives to deferoxamine in
aluminum toxicity. J Clin Toxicol 1988; 26: 67-79.
[37] Schmidt M, Pohlmann U, Golf S, Temme H, Graef V. Roka L,
Riediger H, Neppel H, Brustle A, Bortz C. The effects of a
magnesium supplementation on the energy metabolism in competitive
swimmers. Magnes Res 1989; 2: 296.
[38] Heaton FW, Rayssiguier Y. Magnesium deficiency and
membrane properties. In: Altura BM, Durlach J, Seelig MS, eds
Magnesium In Cellular Processes and Medicine. Basel: Karger,
1987: 121-30.
[39] Johansson BW. Magnesium infusion in decompensated
hypomagnessemis patients. Acta Pharmacol Toxicol (Copen) 1984;
54: 125-8.
[40] Cox IM, Campbell MJ, Dowson D. Red blood cell magnesium
and chronic fatigue syndrome. Lancet 1991; 337: 747-60.
[41] Galland L. Magnesium and inflammatory bowel disease.
Magnesium 1988; 7: 78-83.
[42] Durlach J, Lutfalla G, Poenaru S, Reba A, Henrotte JG,
Fabiani F, de Vernejoul F. Latent tetany and mitral valve
prolapse due to chronic primary magnesium deficit. In: Halpern
Mi, Durlach J, eds. Magnesium Deficiency. Basel: Karger, 1985;
102-12.
[43] Galland L. Magnesium deficiency in mitral valve prolapse.
In: Halpern Mi, Durlach J, eds. Magnesium Deficiency. Basel:
Karger, 1985: 117-19.
[44] Fernandes JS, Pereira T, Alcazar A, Franca A, Andrade R,
Pereira JN, Pessoa J, Rodrigues JC, Guerra L, Laires MJ, Halpern
MJ. Idiopathic mitral valve prolapse and the neuromuscular form
of primary magnesium deficit. In: Halpern MJ, Durlach J. eds
Magnesium Deficiency. Basel: Karger, 1985: 120-21.
[45] Altura BM. Calcium antagonist properties of magnesium:
implications for antimigraine actions. Magnesium 1985; 4:
169-75.
[46] Abraham GE. Primary dysmenorrhea. Clin Obs Gyn 1978; 21:
139.
[47] Cheeseman AJ, Clark JB. Influence of the malate-aspartate
shuttle on oxidative metabolism is a synaptosomes. J Neurochem
1988; 50: 1559-65.
[48] Wiesner RJ, Kreutzer U, Rosen P, Grieshaber MK.
Subcellular distribution of malate-aspartate cycle intermediates
during normoxia and anoxia in the heart. Biochim Biophys Acta
1988; 936: 114-23.
[49] Hoehl C, Oestreich R, Rosen P, Wiesner R, Grieshaber M.
Evidence for succinate production by reduction of fumarate during
hypoxia in isolated adult rat heart cells. Arch Biochem Biophys
1987; 259: 527-35.
[50] Wilson MA, Cascarano J. The energy-yielding oxidation of
NADH by fumarate in submitochondrial particles of rat tissues.
Biochim Biophys Acta 1970; 216: 54-62.
[51] Wiesner RJ, Rosen P, Grieshaber MK. Pathways of succinate
formation and their contribution to improvement of cardiac
function in the hypoxic rat heart. Biochem Med Metab Biol 1988;
40: 19-34.
[52] Dunaev VV, Tishkin VS Milonova NP, Belay IM, Makarenko
AN, Garmash SN. Effect of malic acid salts on physical working
capacity and its restoration after exhausting muscular work.
Farmakol Toksikol 1988; 51(3): 21—25.
[53] Bobyleva-Guarriero V, Lardy HA. The role of malate in
exercise-induced enhancement of mitochondrial respiration. Arch
Biochem Biophys 1986; 245(2): 470-6.
[54] Agbanyo FR, Moses G, Taylor NF. L-malate transport and
proton symport in vesicles prepared from Pseudomonas putida.
Biochem Cell Biol 1986; 64(11): 1190-94.
[55] Belikova YO, Kotlyar AB, Vinogradov AD. Oxidation of
malate by the mitochondrial succinate—ubiquinone reductase.
Biochim Biophys Acta 1988; 936: 1-9.
[56] Carlsen BD, Lambeth DO, Ray PD. Synthesis of malate from
phosphoenolpyruvate by rabbit liver mitochondria: implications
for lipogenesis. Biochim Biophys Acta 1988; 965: 1-8.
[57 Bobyleva-Guarriero V, Battelli D, Bellei M, Lardy HA.
Sources of intramitochondrial malate. FASEB J 1989; 3:
2208-1l.
[58] Kostin VI, Use of malate and NAD for the correction of
blood supply and oxygen balance of the acutely ischemic
myocardium. Farmakol Toksikol 1986; 49(33): 44-6.
[59] Schantz PG, Sjoberg B, Svedenhag J. Malate-aspartate and
aipha-glycerophosphate shuttle enzyme levels in human skeletal
muscle: Methodological considerations and effect, of endurance
training. Acta Physol Scand 1986; 128: 397-407.
[60] Gordon SE, Kraemer WJ, Pedro JG. Increased
acid—base buffering capacity via dietary supple mentation:
anaerobic exercise implications. J Appl Nut 1991; 43: 40-8.
[61] Taroni F, Di Donato S. Purification and properties of
cytosolic malic enzyme from human skeletal muscle. Int J Biochem
1988; 20(8): 857-66.
(62] Abraham GE. The effects of nutrients on premenstrual mood
in nutrients and brain function. Essman WB, ed. Basel: Karger,
1987: 167.
[63] Katsurada A, Iritani N, Fukuda H, Noguchi T, Tanaka T.
Transcriptional and posttranscriptional regulation of malic
enzyme synthesis by insulin and triiodothyronine. Biochim Biophys
Acta 1988; 950: 113-17.
[64] Kobayashi M, Morishita H, Okajima K. Successful treatment
with succinate supplement in a patient with a deficiency of
Complex I (NADH-CoQ reductase). In: The Fourth International
Congress of Inborn Errors of Metabolism 1987: 148.
[65] Lehninger AL. Biochemistry. Worth Publishing, 1978:
411.
[66] Mayes PA. Oxidative phosphorylation and mitochondrial
transport systems. In: Murray RK, ed. Harper’s
Biochemistry. Appleton and Lange, 1990: 112-23.
[67] Goldstein A. Biostatistics. MacMillilan, 1965: 61.
This page was first uploaded to The Magnesium Web Site on July
19, 2002
http://www.mgwater.com/
