MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
Part III: Chapter 11
SKELETAL AND RENAL EFFECTS OF MAGNESIUM DEFICIENCY
11
Magnesium, Bone Wasting, and Mineralization
11.1. Mobilization of Bone Magnesium
Relatively little attention has been paid to the importance of magnesium in bone metabolism, except to the degree that it affects the activity of the parathyroid glands and C cells and their secretion of parathyroid hormone PTH and calcitonin (CT), and the response of target organs. However, experimental magnesium deficiency causes abnormalities in skeletal structure, enzymes, and mineralization that resemble some of those seen in several clinical bone diseases. Depending on the degree and duration of the magnesium deficiency and concomitant dietary or iatrogenic imbalances (of magnesium with calcium, phosphates, vitamin D, and other calcemic agents), the pathologic skeletal findings can range from osteopenias to osteosclerosis. The effects of vitamin D, calcium, and phosphorus on magnesium requirements and on skeletal responses have been intensively studied, particularly in the 1930s, when vitamin D toxicity was the focus of much attention. Many of the results are conflicting, probably due to the dietary variations, and to species differences in requirements (i.e., of vitamin D). Only those portions of the PTH/CT/Mg data that deal directly with magnesium and bone are considered here. Much of that relating to gestational abnormalities has already been discussed. The relatively little information found on heteroionic magnesium/calcium exchange in bone, and on the magnesium interrelationships between the phosphatases that affect mineralization, alkaline phosphatase and pyrophosphatase, are brought into focus as possibly providing some insight into the conflicting and confusing data on mechanisms of pathologic skeletal processes.
Largely disregarded in the treatment of bone disease is the possibility that some of the therapeutic agents (used to increase bone mineralization) might adversely affect bone metabolism by causing loss of skeletal magnesium. Calcium, phosphorus, and vitamin D all increase magnesium requirements; the intakes of all have been rising during this century, while that of magnesium has been falling. Since plasma levels of magnesium are maintained within very narrow limits, even in the face of insufficient intakes or excessive losses, the magnesium is mobilized from the tissue stores. Bone constitutes the largest total source; it contains two-thirds of the total body magnesium (Review: Heaton, 1971). Much of bone magnesium is quite labile, especially in young animals. Were the bone magnesium merely an inert storage depot, this would be a benign means of providing magnesium for the function and structure of life-preserving tissues (e.g., cardiovascular and renal), as well as preventing acute neuromuscular signs of magnesium depletion. For short periods of time, and more in young than in older individuals, availability of bone magnesium probably serves as a safety device that prevents serious systemic signs of magnesium deficiency. However, long-term loss of magnesium from the bone causes disturbances of bone modeling, remodeling, and turnover, with resultant bone abnormalities. Depending upon the supply of the calcemic agents or phosphate, it can give rise to formation of brittle chalky bones or to osteopenia. The mobilized bone constituents contribute to the renal damage of magnesium deficiency.
Because the amount of magnesium bone is only 1/40 to 1/50 that of calcium (Duck- worth et al., 1940), relatively few investigators have given it much consideration as a significant bone mineral, either in bone metabolism or as a source of emergency magnesium supply. Bone magnesium is an important source, especially in young animals (McAleese et al., 1961), an observation supported by the drop in bone magnesium immediately after convulsions of magnesium deficiency (Orent et al., 1934; Martindale and Heaton, 1964). Differences in responses to vitamin D, PTH, and CT influence the mobilization of magnesium during magnesium deficiency and have led to diverse findings. Many of the studies have dealt with the influence of magnesium deficiency and repletion, with high and low calcium, phosphorus, and vitamin D intakes, on metabolic balance. They are not considered here, unless bone values are also given, since positive balances (e.g., of calcium and phosphorus) can be achieved by metastatic calcification, as well as by increased bone mineralization and can occur even with bone demineralization. Also, failure to exhibit negative magnesium balance under conditions that cause abnormal bone structure might be related to the initial shift of bone magnesium and calcium (e.g., the increase in bone magnesium/calcium ratio in rickets).
Some of the disparate findings in the different studies might well be the result of use of widely differing diets in the magnesium deficiency studies: diets that provide 3200 to 8000 parts per million (ppm) of calcium, 1900 to 5100 ppm of phosphorus, and 1150 to 1,000,000 IU of vitamin D per kilogram of diet mix, and 3 to 100 ppm of magnesium (Larvor and Durlach, 1971a). In some of the studies analyzed and tabulated by Larvor and Durlach (1971a), only the magnesium provided was indicated. Thus, the studies cited in the following sections are not strictly comparable.
11.2. Influence of High Vitamin D and High or Low Calcium Intakes
11.2.1. High Calcium: Decreased Mobilization
Most studies of hypervitaminosis D are in rats, which are commonly fed rations rich in calcium and phosphate, as well as in vitamin D. All three of these supplements cause magnesium loss (Reviews: Heaton, 1971; Larvor and Durlach, 1971b; Seelig, 1971). High calcium intakes compete with magnesium for intestinal absorption and renal tubular reabsorption (cited reviews), and high calcium extracellular levels result in exchange of bone magnesium for calcium.
Orent et al. (1934) were the first to note that rats on a low-magnesium, very high-calcium diet (Mg/Ca= 5/ >3000), also fed vitamin D lost about half the original percentage ash magnesium, but doubled the percentage ash calcium in their long bones. The magnesium was 1/3 normal for the same-age control rats. They noted that in rats sacrificed during convulsions, the magnesium level rose in the blood and dropped sharply in the bones, suggesting rapid mobilization of bone magnesium at that time. Nonetheless, the total accretion of bone magnesium exceeded the amount fed, and the authors speculated that it might have derived from organs such as liver, kidney, and heart, and possibly from muscle, organs which also increased in calcium content. They suggested that lowering of the skeletal magnesium ratio might have been caused by their having added vitamin D to the rats' rations. Comparable reduction in bone magnesium was reported by Cunningham (1936b) in rats fed the same magnesium-deficient diet (Kruse et al., 1932). Watchorn and McCance (1937) provided cod liver oil rather than viosterol to the rats that they maintained on a subacute magnesium-deficient regimen for up to three months. Notable were renal calcification and hepatic and skeletal damage. The long bones and teeth were brittle, and the teeth were loose in their sockets. Even though few of the many studies of vitamin D toxicity (which emphasized renal and cardiovascular damage) provided magnesium values, some of the findings (which subsequent work suggests might have been contributed to by magnesium depletion caused by the regimens) are included here. For example, rats developed overcalcification of bones and teeth (which is suggestive of a process that inhibits mobilization of bone minerals) when they were given high-dosage vitamin D, as well as diets rich in calcium and phosphorus (L. J. Harris, 1932; Shelling and Asher, 1932). In the late stage of moderate hypervitaminosis D, or with very high doses, there were cessation of osteogenesis and bands of less calcified bone near the epiphyses. (The histological changes described are much like those reported in magnesium-deficient rats and in human osteopetrosis.) Storey (1960) noted that intermittent hypervitaminosis D produces similar lesions. Comparable hypercalcification of bones, which lost 74% of control magnesium content, was found in magnesium-deficient chicks supplemented with calcium and vitamin D (C. Reddy et al., 1973). They also had increased unmineralized osteoid and cortical thickening, that was reversed rapidly on magnesium repletion. A recent study with hypervitaminosis D in pigs clarified the nature of the bone pathology with increasing doses. At 5 and 25 times the recommended dose there was osteopetrosis; at higher doses there were hypercalcemia and hypophosphatasia (Chineme et al., 1976).
On the other hand, it was suggested that rats that developed hypomagnesemia during their overdosage with vitamin D, and that did not exhibit hypermagnesuria, might be depositing magnesium in their bones (Richardson and Welt, 1965). Wallach et al. (1966) confirmed this premise in dogs on 1% dietary calcium intake, given very high vitamin D doses, that became hypercalcemic and hypomagnesemic. Their bones had only slightly increased total calcium and moderately increased (p < 0.2) exchangeable calcium. Their total bone magnesium, however, had increased significantly (p < 0.001), but there was little change in the exchangeable magnesium content.
The total bone mineral distribution of the dogs given short-term toxic doses of vitamin D (Wallach et al., 1966) resembles that reported in the early rickets studies in rats [Malcolm, 1904; Mellanby, 1926 (cited by McHargue and Roy, 1930)]. Since these animals were hypomagnesemic, as were rats overdosed with vitamin D (Hanna, 1961a; Harrison and Harrison, 1964), it can be speculated that they were in the early stage of development of vitamin-D-resistant rickets (i.e., hypervitaminosis D rickets: Ham and Lewis, 1934). Longer-term hypervitaminosis D plus high calcium intakes, as in the Watchorn and McCance (1937) and Storey (1960) studies, might be experimental models of infantile hypercalcemia, which is associated with osteosclerosis as well as with metastatic calcification (Review: Seelig, 1969b).
Despite the magnesium loss caused by the vitamin D and calcium excesses, caution must be exercised in repleting the magnesium. Whittier and Freeman (1971) have demonstrated that metastatic calcification has been potentiated by giving magnesium to rats with hypercalcemia caused by hypervitaminosis D. This recalls the speculation that the use of magnesium laxatives, to manage the obstipation of hypercalcemic children, might have contributed to their metastatic calcification (Creery, 1953; Lowe et al., 1954; Review: Forfar thesis). The rationale for this paradoxical observation is considered elsewhere in this chapter. It is important to keep in mind now that hypophosphatemic rickets, refractory to high dosage vitamin D and calcium, has been reported to be responsive to magnesium.
Fetal and neonatal spontaneous fractures and lesions resembling those of osteogenesis imperfecta and hypophosphatasia develop in pups of rats given high doses of vitamin D and in infants born with intrauterine growth retardation, both conditions that might be related to fetal magnesium deficiency.
Early or acute magnesium deficiency has been shown to stimulate PTH secretion, but the concomitant hypercalcemia in the experimental model and most clinical conditions in which hypervitaminosis D plus high calcium intakes play a role would function to decrease PTH secretion, outweighing the stimulant effect of magnesium deficiency. Additionally, early and acute magnesium deficiency has stimulated CT secretion, an effect enhanced by hypercalcemia (Stachura and Pearse, 1970). Thus, the overall effect on bone of diets low in magnesium and high in calcemic agents is decreased mobilization of bone calcium, with replacement of surface bone magnesium by calcium.
11.2.2. Low Calcium: Increased Mobilization
Rats on normal diets given high-dosage vitamin D without calcium supplements or low-calcium diets were shown, in early studies, to exhibit resorption of compact bone, an effect attributed to vitamin-D-induced bone mineral mobilization (Duguid. 1930a; L. J. Harris, 1932; Shelling and Asher, 1932). In a 1953 review, Nicolaysen and Eeg-Larsen reported that the dominant feature of hypervitaminosis D is dissolution of formed bone and dense calcification of hypertrophic cartilage.
Duckworth et al. (1940), whose magnesium-deficient rats had much less bone calcification than did those of Orent et al. (1934), did not list vitamin D as a dietary constituent. They found that weanling rats, kept on a diet adequate in calcium but low enough in magnesium to result in tetany or convulsions and death by 6 days to a month, had less growth and markedly less magnesium (percent in ash) in their bones than did littermates on the same diet but supplemented with magnesium. In contrast, the magnesium-deficient rats had no decrease (percent in ash) of calcium or phosphorus. In fact, they had a slightly increased percentage of bone ash calcium. Those on the deficient diet for 16 and 23 days exhibited the greatest percentage loss of magnesium as compared to adequately pair-fed rats (0.39 → 0.34% versus 0.83 → 0.74% Mg in bone ash). Rapid replenishment of the bone magnesium was exhibited by rats fed deficient diets for 6 days and then adequate diets for 10 days. The bones of the rats that survived the magnesium-deficient period had more fragile bones than did those reared on adequate rations, and give histologic evidence of abnormal matrix. They then found that rats fed diets deficient in both calcium and magnesium survived longer than did those fed diets adequate in calcium but low in magnesium (Duckworth and Godden, 1941). The rats low in both cations more quickly mobilized more magnesium from their bones, a possible explanation of their longer survival. The rate of bone growth determined the amount of the magnesium that could be liberated because of the demand of the skeleton itself for magnesium. They then showed that when the diet was free of calcium but contained no less than 6 ppm of magnesium, the demineralized bone ash contained progressively more magnesium and less calcium (Duckworth and Godden, 1943). Thus, to a limited extent, the magnesium replaced calcium in the bone crystal. This did not occur with deficiency of both cations.
The mobilization of bone mineral (particularly calcium, the major bone mineral, but also magnesium) by excess vitamin D with low calcium and magnesium intakes or body reserves might be a direct effect, as has been shown with vitamin D metabolites (Trammel et al., 1969; Raisz et al., 1972; Reviews: Norman and Henry, 1974; Norman et al., 1975/1977; DeLuca, 1976) or one that is mediated by secondary hyperparathyroidism. That hypocalcemia causes increased PTH secretion is well established. The effect of hypomagnesemia is neither as well known nor as clear-cut. Larvor et al. (1964a) demonstrated that magnesium deficiency (in a calf on normal calcium and vitamin D intakes) caused hyperplasia and osteitis fibrosa. Indirect evidence of increased PTH secretion in rats on diets low in magnesium but adequate in calcium was provided by investigators who prevented hypercalcemia in magnesium-deficient rats by parathyroidectomy (Kukolj et al., 1965; Gitelman et al., 1965, 1968b). I. Clark (1969b) provided evidence that magnesium deficiency in rats fed adequate calcium and phosphate exerts a slight stimulant effect on PTH secretion.
In vitro studies have provided direct evidence of the PTH secretory effect of magnesium deficiency. Perfusion of the parathyroids of goats and sheep (which are separate from their thyroids), with hypomagnesemic, normocalcemic solution resulted in increased PTH secretion (Care et al., 1966; Buckle et al., 1968), an effect that was verified by Sherwood (1970) and his colleagues (Sherwood et al., 1970, 1972; Targovnik et al., 1971). Despite this clear laboratory evidence, severe clinical magnesium deficiency has been shown to cause relative parathyroid failure (Muldowney et al., 1970; Anast et al., 1972, 1976; Anast, 1977; Suh et al., 1971, 1973; L. Chase et al., 1974; Avioli, 1978), an effect that can be mediated by decreased PTH release (Anast, 1977) or skeletal unresponsiveness (Estep et al., 1969; C. Reddy et al., 1973; Levi et al., 1974; Medalle et al., 1973, 1976). However mediated, Forbes and Parker (1976/1980) have shown diminished bone resorption (as measured by 45 levels) in magnesium-deficient young rats.
Why a condition associated with increased PTH secretion (that mobilizes bone minerals and leads ultimately to magnesium loss, as well as hypercalcemia) should be associated with increased levels of bone magnesium in the acute studies, is difficult to explain. It is conceivable that the enhancement by PTH of mitochondrial uptake of magnesium (Rassmussen et al., 1964) might be contributory. The increase in bone magnesium, associated with hypervitaminosis D, might be correlated with a possible PTH-mediated early bone uptake of magnesium. Since magnesium participates in osteoblastic activity and osteoid formation, the net result of the imbalance produced by concomitant hypervitaminosis D and low calcium intake (and that causes hypomagnesemia) might well be the high magnesium/calcium bone ratio, and the relative excess of osteoid, such as is seen in clinical rickets and in hyperparathyroidism. It might also include the osteomalacia of malabsorption syndromes and vitamin-D-resistant rickets following high-dosage calcemic therapy.
Possibly the initial response to hypomagnesemia of the CT producing C cells is increased secretion, even in the absence of hypercalcemia (Rojo-Ortega et al., 1971). It is conceivable that this response functions to inhibit release of bone magnesium, as well as to partially counteract the mobilization of bone calcium of animals loaded with vitamin D. However, compensatory CT secretion is insufficient to counteract calcium mobilization from bones of rats given very high doses of vitamin D (Mittleman et al., 1967).
Despite the (possible) increase in CT secretion, hypervitaminosis D (usually in adults whose calcium intake is not high) has caused hypercalcemia and bone demineralization, as well as metastatic calcification.
11.3. High Phosphate Intakes: Effects on Bones
11.3.1. Effects on Bone Magnesium
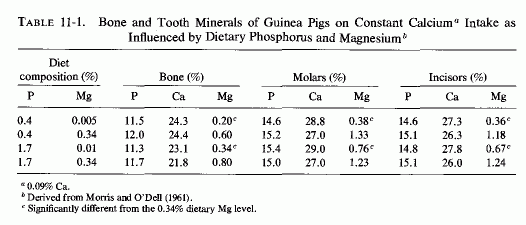
E. R. Morris and O'Dell and their colleagues studied the influence of increasing the phosphate intake on skeletal and dental structures of guinea pigs on low to normal magnesium intakes, keeping the calcium intake adequate and constant (O'Dell et al., 1960; E. R. Morriss and O'Dell, 1961). Although the calcium and phosphorus levels of the hard structures remained essentially the same in magnesium-deficient and control animals, regardless of the intake of phosphate, the animals on a diet low in both magnesium and phosphate had one-third as much magnesium in their bones and teeth as did animals on control magnesium intakes, also low in phosphate. Increasing the phosphate increased the magnesium requirements for survival, and induced changes in bone and tooth minerals (O'Dell et al., 1960). At both the low- and high-phosphate intakes, increasing the magnesium levels 70- and 35-fold, respectively, significantly increased the magnesium levels of the hard structures (Table 11-1) and prevented their structural defects. The investigators speculated that the phosphate-induced loss of skeletal magnesium caused abnormalities in the matrix. Forbes (1961) evaluated the effects of varying dietary ratios of calcium, magnesium, and phosphorus in weanling rats. He demonstrated that on marginal magnesium intakes, overt magnesium deficiency was produced only when excesses of both calcium and phosphorus were provided. The percentage of magnesium in femur ash was lowest in magnesium-deficient rats supplemented with both calcium and phosphorus and was almost as low when supplemented only with phosphorus (Forbes, 1963).
In studies of the effects of magnesium depletion and repletion on rats depleted of or provided adequate calcium and phosphorus, I. Clark (1966, 1968, 1969a, b, 1971/1973, 1977) showed that the amount of each ion required or tolerated is influenced by the intakes of the others (Fig. 11-1). He also showed that femoral weight and calcification is depressed without optimal magnesium intake. In a study of bone minerals in rats on constant calcium and phosphate intakes, but on low-to-high magnesium supplements, Clark and Bélanger (1967) found declining bone calcium and magnesium as the dietary magnesium-to-calcium ratio declined. Meyer and Busse (1976/1980) reported that changing vitamin D intakes did not alter bone-magnesium levels in rats on high phosphorus intakes, although they confirmed that vitamin D slightly lowered blood levels of magnesium. They found that the magnesium-bone content of rats fed diets with slightly higher phosphorus than calcium content was slightly higher than that of rats fed diets with three times as much calcium as phosphorus. In sheep, there was also more magnesium in bone ash than when the dietary calcium to phosphorus ratio was low than when it was high.
11.3.2. High P/Ca; P/Mg and Bone Wasting; Mineralization
11.3.2.1. Bone Wasting
In view of the cited evidence that excess phosphate decreases bone magnesium levels, and the importance of magnesium in maintaining normal bone metabolism, the evidence that experimental high phosphorus/calcium ratios causes bone wasting is relevant. Data referable specifically to the renal calcinosis produced by diets with high phosphorus/magnesium ratios, or that cause increased phosphorus mobilization, will be discussed in Chapter 13..
Shelling and Asher (1932) studied the influence of different proportions of dietary calcium and phosphorus on bone and soft tissue calcification of rats given no vitamin D, or given moderately high to very high doses. Young rats on high P/Ca dietary ratios developed osteoporosis, which was intensified by increasing the phosphorus intake further, and further worsened by addition of large doses of vitamin D. Microscopic studies of young rats on low calcium/high phosphorus and vitamin D (40,000 times the antirachitic dose) showed a progressive decrease in the number of trabeculae with the duration of the experiment. At the end (by the 26th day), the trabeculae had been replaced by remnants of osteoid, osteoblasts, and tiny fragments of calcified material. The similarity of these abnormalities to those seen in the genetic abnormality, hypophosphatasia, and the low alkaline phosphatase levels of infants with hyperreactivity to vitamin D deserves consideration.
More recently, the risk of bone wasting (caused by high P/Ca in the diet) has been studied by Krook et al. (1971, 1975). They demonstrated nutritional osteoporosis in dogs, horses, pigs, and monkeys kept on diets with high phosphorus/calcium ratios for prolonged periods of time. The disease is characterized by hypercalcemia and hypophosphatasia; the bone damage, both in long bone and in mandibles, is related to secondary hyperparathyroidism (attributed both to the low dietary calcium and high dietary phosphate). The histological changes resemble those of osteoporosis, and are characterized by loss of matrix and by demineralization, particularly in the subperiosteal areas of the compact bone. In trabecular bone, osteocytic osteolysis occurs in the center, and the trabeculae become thinner. It is likely that excess phosphate-induced depletion of magnesium contributes to the enzyme, parathyroid, and bone changes.
Feinblatt et al. (1970) also observed that high phosphate/calcium ratios (in rats) cause similar lesions. They also demonstrated that phosphate infusions reduced the hypercalcemia caused by PTH administration, but did not alter its increase of urinary hydroxyproline secretion. Thus, their findings indicate that phosphate does not block bone resorption, but they assume that it increases bone mineralization. Comparable osteoporotic changes have been produced by excess dietary phosphate in adult intact and parathyroidectomized rats (G. Anderson and Draper, 1972) and in aging mice (Krishnarao and Draper, 1972) and rats (Draper et al., 1972). Lutwak (1974) has commented that such intakes are common in the American diet, and suggested that they might be contributory to the high frequency of osteoporosis and periodontal disease. In contrast, Berlyne et al. (1973b) have attributed the rarity of renal osteodystrophy in Israel to a low phosphorus intake.
11.3.2.2. Bone Mineralization
That phosphate loads might increase bone mineralization was first proposed by
F. Albright et al. (1932), who speculated that inorganic phosphate's antihypercalcemic effect (in hyperparathyroidism) was mediated by inhibition of bone resorption. Raisz and Niemann (1969) demonstrated this effect in vitro, and then showed that increasing the phosphate concentration of suspending media stimulated collagen synthesis by rat bone (Raisz, 1970). However, phosphate loading has stimulated PTH secretion, and failed to inhibit PTH-induced bone resorption, as indicated by continued excretion of bone minerals and hydroxyproline (Pechet et al., 1967; Feinblatt et al., 1970; Rasmussen et al., 1970). Despite its failure to suppress PTH-bone resorption, Pechet et al. (1967) reported that the neutral phosphate stimulated bone formation and mineralization. They explained this finding on the basis that considerable amounts of phosphate are bound by collagen and initiate crystal nucleation and growth (Glimcher and Krane, 1964).
11.4. Influence of Metabolic Activity of Bone on Availability of Bone Magnesium
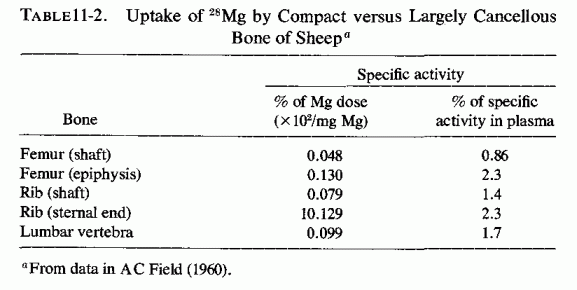
Availability of radioisotopes of magnesium (i.e., 28Mg) has permitted study of the influence of the metabolic activity of bone on its uptake and release of magnesium during short periods of time. Brandt et al. (1958) found considerable variability in skeletal 28Mg uptake by different bones of different dogs, and postulated that the rate of uptake is likely to be affected by many factors: growth, renal function, and the magnesium stores of the body. A. C. Field (1960) found that there was marked variation in magnesium uptake from bone to bone in sheep. It was greater in regions of rapid bone metabolism than in compact bone (Table 11-2). Using a radiographic procedure to measure the uptake of 28Mg in puppies, Glaser and Gibbs (1962) showed that the growing, actively metabolizing portion of bone (the epiphyseal line) concentrates most of the 28Mg that is taken up by bone, as compared with the diaphysis, the least active portion.
In a study comparing predominantly the influence of age on the amount of magnesium mobilized from bone of magnesium-deficient rats (B. S. W. Smith and Field, 1963), there was relatively more magnesium lost per unit of mandible than per unit of femur. More magnesium was lost from the bones of the young rats (mandibular versus femoral magnesium loss = 33.3% versus 28.2%), but proportionally more was lost from the mandibles of the older rats (13.4% versus 9.5%). Parr (1957) confirmed the greater loss of magnesium from cancellous than from compact bone of magnesium-low calves. McAleese (1961) showed that epiphyses of magnesium lambs took up more 28Mg than did the diaphyses, indicating either more magnesium loss from the area of bone growth, its greater magnesium requirement, or both.
In a serial study of loss of magnesium from vertebrae, R. H. Smith (1959) amputated the terminal caudal vertebra at monthly intervals from magnesium-deficient and control calves, and found that the magnesium content of the bone ash dropped before the appearance of clinical signs of deficiency. Larvor et al. (1964a) showed that the diaphyses of magnesium-deficient calves lost less magnesium (compared with controls) than did the vertebrae. The ratio of vertebral magnesium in deficient versus control calves was 0.16:0.35; that of diaphyseal magnesium was 0.25:0.41. There was very little difference in bone calcium or phosphorus in the magnesium-deficient and control calves. B. S. W. Smith and Field (1963) found that magnesium-deficient rats lost relatively more magnesium from mandibular than from femoral bone. Minimal osteoblastic and alkaline phosphatase activity was found in alveolar bone of magnesium deficient rats (Trowbridge and Seltzer, 1967).
Aikawa (1965) demonstrated that the rate of bone uptake of 28Mg is influenced by the metabolic activity of the bone cells. Administration of insulin and glucose (Aikawa, 1960a) or of pyridoxine (Aikawa, 1960c) increased the bone uptake of 28Mg inhibitors of thyroid function of pyridoxine activity, or irradiation, decreased bone 28Mg uptake (Aikawa, 1960b; Aikawa and Reardon, 1963; Aikawa, 1965). MacManus and Heaton (1970) demonstrated that, in vitro, metabolically active bones release more magnesium to a magnesium-free medium than do bones whose enzymatic activity has been destroyed by aging. Heaton (1971) thus concludes that magnesium is released by a mechanism that is dependent on the metabolic activity of bone cells. (In the in vitro system, most of the magnesium released reflects establishment of a physicochemical equilibrium between the bone and its surrounding fluid.)
Bones with a high proportion of cancellous to compact bone (more metabolically active) develop clinically manifest osteopenia before predominantly compact (long) bones do. Thus, the greater loss of magnesium from such bones, relatively early in magnesium deficiency, might be clinically important, in view of magnesium's significance in so many enzyme systems (Reviews: Lehninger, 1950; Green and MacLennan, 1960; Heaton, 1976/1980). The intensification of magnesium deficiency by calcemic agents and phosphates, such as are commonly used in treatment of osteopenias and hypercalcemic states, might intensify bone matrix abnormalities and lead to the formation of hypermineralized bones with little matrix. It is possible such bones are similar to the brittle chalky bones and teeth seen in magnesium-deficient rats fed diets high in calcium, phosphate, and vitamin D Animals fed magnesium-deficient rations more similar to the human diet (high P/Mg and P/Ca) ratios tend to develop osteopenia. Rarely is the possibility that calcemic treatment of clinical osteopenias might intensify magnesium deficiency considered (Amiot et al., 1969; Durlach, 1971).
11.5. Influence of Age on Mobilization of Bone Magnesium
That skeletal magnesium is not readily mobilized in adult animals was suggested by the early work of Cunningham (1936a), who showed that bones from lactating cows with grass tetany and hypomagnesemia contained normal amounts of magnesium. Calves, however, kept on a diet low enough in magnesium (Mg/Ca=¼) to cause convulsions or death in 8 to 16 days, lost about two-thirds of their bone magnesium (Blaxter et al., 1954). Blaxter (1956) later evaluated the tissue magnesium changes in magnesium-deficient calves and found that soft tissue levels were not significantly depleted, but that there was a 56% loss of bone magnesium. His data suggested that the loss of skeletal magnesium takes place at the surface of the bone crystals, and that it occurs more readily in young than in old animals. Less severely depleted calves lost less bone magnesium, but more than did cows with lactation tetany (Parr, 1957), a condition associated with magnesium depletion.
In the case of rats, which continue to grow after they reach sexual maturity, the results are generally not as clear-cut. Breibart et al. (1960) found that young rats (20 to 30 days of age:44-100 g) exchanged 31-46% of their bone magnesium with 28Mg whereas 60 to 180 day-old rats (130-225 g) exchanged only 4-5% of bone magnesium. Young rats (90-110 g) that were kept on a magnesium-deficient diet high in calcium (Mg/Ca = 3.8/1500 mg/100 g diet) that maintained their growth, but at 1/6 the control rate, showed a pattern of distribution of injected 28Mg different from controls (Chutkow, 1965). Initially (within 3 minutes after the injection) there was prompt uptake of greater amounts of 28Mg (than in controls) by all tissues, including bone. Thereafter, most of the 28Mg was diverted to the soft tissues; the skeletal uptake of 28Mg did not exceed that achieved during the first few minutes. The study of A. C. Field and Smith (1964) was on the effect of magnesium deficiency on the uptake of 28Mg by mature rats (9-12 months old; avenging 400 g in weight), but cannot be directly compared with the Chutkow study (1965) because the Mg/Ca ratio was much lower: CaCO3 : 75 parts, versus hydrated MgSO4 : 26 parts in controls, and absent from deficiency diets. They (Field and Smith) found that the bones of magnesium deficient rats took up less 28Mg than did the viscera (versus controls). The mandible took up relatively more magnesium than did the femur, the uptake of which was about equal to that of skeletal muscle. The relative specific activities (the ratio of that of the tissue to that of plasma, a measure of the proportions of exchangeable magnesium) of bone from the magnesium-deficient adult rats were less than in control rats, in contrast to the relative specific activities of the vital organs.
B. S. W. Smith and Field (1963) compared the amount of magnesium mobilized from the bones of 8-week-old male and female magnesium-deficient rats (180 and 140 g) with that from 9-to 12-month-old males (average weight: 400 g). They found that the young rats lost much more bone magnesium than did the old rats. The femurs of the magnesium-deficient young rats showed 28.2% magnesium depletion from femurs, as compared with controls; the mandibles showed somewhat more: 33.3% magnesium depletion versus controls. There was less loss of magnesium from the adult rats: 9.5% depletion in femurs; 13.4% depletion in mandibles versus controls. Martindale and Heaton (1964), however, found that mature rats, 4 to 5 months of age, lost bone magnesium rapidly during the first 15 days of deficiency, and then more slowly to reach about half the starting value after 62 days. The pattern of change was similar to that seen in blood plasma. These rats showed a significant rise in bone content of calcium and sodium, a finding in accord with the early studies (Orent et al., 1934), in which rats were given rations high in calcium. [Note that most magnesium-deficient rat diets are high in calcium, phosphate, and vitamin D (Review: Larvor and Durlach, 1971b).]
11.6. Physicochemical Exchange of Bone Magnesium and Calcium
The hypocalcemia of severe magnesium depletion, which has been attributed to target organ unresponsiveness to PTH (or to failure of PTH release or secretion), has been explained by physicochemical factors involving ionic exchange of magnesium and calcium at the bone surface. Heaton (1971) has reviewed the evidence that bone magnesium is much more readily available than is bone calcium. (About a third occurs within the apatite crystals, the remainder being either adsorbed on the crystal surface or present in solution within the hydration shell around the crystals.) Duckworth and Godden (1941) showed that calcium exchanges for magnesium in the apatite crystal during magnesium depletion. Neuman and Neuman (1957) suggested that calcium ions can enter the extracellular fluid from bone only if the bone crystal takes up other cations (i.e., magnesium) to maintain electroneutrality. R. H. Smith (1961) speculated that the correlation of falls in plasma magnesium and calcium in magnesium-deficient calves might affect the availability of bone calcium. He observed that the fall in bone magnesium levels reflects that of serum magnesium, and that thus there is less extracellular magnesium available for exchange with calcium. Zimmet (1968) considered this possibility in interpreting the hypocalcemia of his magnesium-depleted patients, noting that Heaton and Fourman (1965) had suggested that magnesium deficiency interferes with release of calcium from bone. Larvor et al. (1964) showed that, during the early stage of magnesium deficiency in the calf, there is a slowing of the rate at which skeletal calcium exchanges with that in the blood. The postulate of Neuman and Neuman (1957) was proved when it was shown that addition of magnesium to an incubation medium increases the release of calcium from bone (Pak and Diller, 1969; MacManus and Heaton, 1970). The magnesium-induced release of calcium is accompanied by liberation of hydroxyproline (MacManus and Heaton, 1970), suggesting that magnesium is involved in bone turnover (Heaton, 1971).
11.7. Alkaline and Pyrophosphatases, Magnesium, and Mineralization of Bone
11.7.1. Magnesium Requirement for Phosphatase Activation and Synthesis
In a 1950 review, Lehninger reported that virtually all phosphatases or phosphate-transferring enzymes are activated by magnesium. As early as 1931, Von Euler and Rydbom found that magnesium, fed to rats on a rachitic diet, increased their subnormal serum phosphatase levels. Snyder and Tweedy (1942) reported that severe experimental magnesium deficiency causes reduced serum alkaline phosphatase activity, an effect that has been verified in cattle and rodents (Larvor et al., 1964a; Heaton, 1965; Pimstone et al., 1966; Trowbridge and Seltzer, 1967; B. Smith and Nisbet, 1968; Hamuro, 1971; Elin et al., l971b; Loveless and Heaton, 1976). The observations that serum and skeletal alkaline phosphatase levels are low in acutely magnesium-deficient rats, and that addition of exogenous magnesium to the medium does not raise the enzyme level to that found in tissues of control rats, indicate that magnesium deficiency reduces the amount of phosphatase present, and not just its activity (Loveless and Heaton, 1976). Low bone levels of alkaline phosphatase have also been found in acutely magnesium-deficient rats by Trowbridge and Seltzer (1967) and Lai et al. (1975). Subacute magnesium deficiency in rats did not cause lowering of bone or serum alkaline phosphatase (Watchorn and McCance, 1937).
In a long-term magnesium depletion study (in patients who had undergone radical face and neck surgery for cancer), serum alkaline phosphatase levels gradually declined (to 1-2 Bodansky units) and did not increase with magnesium supplementation until the 56th day of repletion (Shils, 1969a). A shorter (1 month) study of healthy young men on a low-magnesium diet showed no reduction in serum alkaline phosphatase, even though their magnesium deficit was demonstrable by retention of large amounts of magnesium during repletion (Dunn and Walser, 1966). These volunteers did not develop hypomagnesemia; it seems likely that their bone stores of magnesium were sufficient to prevent interference with serum alkaline phosphatase activity. Possibly masking a (presumed) decrease in enzyme synthesis might be mobilization of alkaline phosphatase from the bone, to a lesser degree than that seen in neoplastic and bone diseases (Taswell and Jeffers, 1963; Moses and Spencer, 1963).
Low serum alkaline phosphatase activity was demonstrated in children with protein calorie malnutrition (R. Schwartz, 1956), a condition in which magnesium depletion has been identified. R. Schwartz (1956) has proposed that the very low serum alkaline phosphatase activity of such children can be correlated with decreased osteoblastic activity. Addition of magnesium to their serum increased the enzymatic activity, but not to the level found in normal children, an effect similar to that reported in studies of magnesium-deficient rats (Heaton 1965; Pimstone et al., 1966).
Low levels of serum alkaline phosphatase have also been found in adults with severe, long-term magnesium depletion (Hanna et al., 1960; Hanna, 1961b; Zimmet et al., 1968; Sutton, 1968; Muldowney et al., 1970; T. B. Connor et al., 1972), and have risen with magnesium infusions (Zimmet et al., 1968). They have also been reported in infants with hypercalcemia related to hypervitaminosis D and in other conditions associated with hypercalcemia (N. J. David et al., 1962). Since both excess vitamin D and calcium predispose to magnesium deficiency, the low alkaline phosphatase levels found in such patients might reflect a conditioned magnesium deficiency. Patients with bone involvement of neoplastic disease (who had hypecalcemia) had lower alkaline phosphatase levels than did those with normocalcemia (Moses and Spencer, 1963). In fact, the hypercalcemia preceded the lowering of enzyme levels (Griboff et al., 1954), possibly a reflection of calcium inhibition of phosphatase.
The genetic bone disorders associated with hypophosphatasia, and in which abnormal magnesium metabolism might play a role, are discussed elsewhere. One such disease, osteosclerosis, which is seen in infantile hypercalcemia [associated with hyperreactivity to vitamin D (Review: Seelig, 1969b) has been duplicated in pigs given 5 to 25 the antirachitic dose of vitamin D (Chinemene et al., 1976)]. On higher doses, the pigs developed hypophosphatasia. The few studies of magnesium in infants with the established syndrome have yielded conflicting results. However, one valuable study has been found that provides evidence suggestive of magnesium malabsorption in an infant with osteopetrosis, who had biochemical findings of hypophosphatemic rickets before high-dosage vitamin D therapy had been started, and whose alkaline phosphatase levels dropped from high to low during the eight months of vitamin D therapy (Pincus et al., 1947). A woman with magnesium-deficient latent tetany and rapidly progressive osteoporosis (Seelig et al., 1975), which was found due to renal magnesium wasting (Seelig et al., 1978), exhibited a sharp drop in her serum alkaline phosphatase following a period of supplementation with 25-OH-D3 during which her serum magnesium level fell further (unpublished data).
Another nutritional imbalance that has caused hypophosphatasia in several species, in association with hypercalcemia, is a normal calcium intake with three to four times as much phosphorus or more (Krook et al., 1975). This diet is considered one that causes nutritional secondary hyperparathyroidism and that is associated with progressive osteopenia. Not considered as a factor in this model is the magnesium deficit that is produced by phosphate loading. It is conceivable that the secondary hyperparathyroidism, the osteopenia, and the hypophosphatasia might all reflect magnesium depletion. Hamuro (1971) reported that on the first day of a high-phosphate, low-magnesium diet there was a slight increase in serum alkaline phosphatase levels in genetically diabetic mice. By days 4 to 6, the enzyme levels dropped to half the initial value. This decrease was not seen when the diet was supplemented with magnesium or when the phosphorus intake was reduced.
Pyrophosphatase, which also has an absolute and relatively high magnesium requirement (Magana et al., 1955; Kunitz and Robbins, 1966) was studied in erythrocytes of magnesium-deficient rats (Elin et al., 1971b). It took two weeks of a diet low in magnesium for red cell pyrophosphatase to drop and two weeks of repletion for it to return to control values. The serum alkaline phosphatase levels dropped more rapidly with magnesium deficiency and responded more quickly with repletion. The authors commented that the delay in pyrophosphatase response to magnesium deficiency and repletion is consistent with the slow fall in erythrocyte magnesium levels with its deficiency (Tufts and Greenberg, 1937) and the evidence that the amount of magnesium in the red cells reflects the magnesium status during their formation (Ginsberg et al., 1962). Heaton (1978) has surveyed the interrelations of magnesium with alkaline phosphatase, pyrophosphatase, and orthophosphatase activities. He has considered the controversy as to whether magnesium inhibits or activates pyrophosphatase activity and concluded that the experimental conditions influence the response of the enzymes to magnesium. The general view is now that magnesium stimulates the hydrolysis of pyrophosphate under normal conditions.
It is difficult to obtain precise data as to phosphatase levels, clinically, since the clinical chemistry laboratories report a single serum alkaline phosphatase figure, not distinguishing between that of skeletal and other (e.g., hepatic) origin. Several fractions have been differentiated (Keiding, 1959; Taswell and Jeffers, 1963). Where there is a disease that is likely to cause magnesium loss, and thus abnormal skeletal alkaline phosphatase activity, the high hepatic alkaline phosphatase values that derive from hepatic damage would obscure skeletal hypophosphatasia. Only research laboratories provide data on the differential alkaline phosphatase levels, and only rarely are pyrophosphatase levels obtained.
11.7.2. Alkaline Phosphatase and Skeletal Mineralization
Robison (1923) postulated that bone alkaline phosphatase liberates inorganic phosphate from organic phosphates, with resultant localized increase in phosphate, which then precipitates the calcium. The in vitro studies that show that considerable amounts of phosphate are bound by collagen and initiate calcium crystallization (Glimcher and Krane, 1964) support the premise that interaction of phosphate with collagen plays a role in bone mineralization (Pechet et al., 1967). During bone growth and during osteolytic processes, the serum alkaline phosphatase activity increases (Griboff et al., 1954; Keiding, 1959). Possibly during new bone formation this reflects increased enzyme synthesis; during bone breakdown it might reflect increased enzyme release. On the other hand, both organic and inorganic polyphosphates inhibit calcium phosphate nucleation and precipitation (in collagen or bone matrix). Without an optimal amount of alkaline phosphatase to destroy the inhibitor, bone mineralization is impeded (Fleisch and Newman, 1961, Fleisch and Bisaz, 1962a,b). Subnormal synthesis or activation of enzymes that act to increase the mineralization process, by removing polyphosphate or pyrophosphate inhibitors, can be correlated with clinical conditions associated with abnormal bone formation and low phosphate levels. The most obvious condition is the uncommon genetic defect, hypophosphatasia, in which the magnesium status has not been explored, but that is characterized by unexplained convulsions in infancy not unlike those of hypomagnesemia, with and without hypocalcemia.
The abnormal high pyrophosphate levels found in serum and bone of infants and children with osteogenesis imperfecta, and the in vitro lowering of their bone biopsies' pyrophosphate content by addition of pyrophosphatase and magnesium suggest that skeletal hypopyrophosphatasia is likely to be an important factor in this disorder. The lowering of serum and urine pyrophosphates of such patients, with magnesium therapy, suggests that abnormal magnesium metabolism (possibly magnesium malabsorption or wasting) might be contributory.
Patients with bone disease, characterized by increased bone turnover (metastatic malignancy, hyperparathyroidism, hyperthyroidism, and Paget's disease) have all exhibited significantly increased urinary outputs of pyrophosphates, as well as of hydroxyproline. This increased pyrophosphate output might be an index of the amount of bone "metabolized" daily (Avioli et al., 1965). Considering this finding and the preliminary evidence that pyrophosphatase might be part of a control mechanism in both formation and resorption of bone, Tenenhouse and Rasmussen (1968) studied its activity in cell suspensions at a fixed physiologic magnesium concentration, at physiologic pH, and as influenced by PTH and CT. They found that PTH inhibits pyrophosphatase activity, and that CT reverses the inhibitory effect of PTH, effects that they considered to be mediated in part by altering the extracellular ionic environment. Orimo et al. (1970) demonstrated that CT administration to rats rapidly increases alkaline pyrophosphatase activity of bone, and suggested that it stimulates bone formation by removing the inhibiting pyrophosphate. These observations should be considered in light of the influence of magnesium on the secretion of both hormones, and on the response of target organs such as bone. It should be kept in mind here that the effects of magnesium deficiency on the hormones and bone depend on the duration and extent of the deficiency. Acute short-term magnesium deficiency increases PTH secretion. Long-term chronic deficiency decreases PTH release and bone response. High-dosage magnesium suppresses PTH secretion. The secretion of CT [which increases osteoblastic activity and decreases bone mineral mobilization (Review: S. P. Nielsen, 1974)] is stimulated by a low magnesium/calcium dietary ratio (Stachura and Pearse, 1970; Rojo-Ortega et al., 1971/1973) and by increased magnesium levels in vitro (Radde et al., 1968, 1970) and in vivo (Care et al., 1971; S. P. Nielsen 1971/1973; S. P. Nielsen and Jorgensen, 1972; Littledike and Arnaud, 1971).
Increased alkaline phosphatase activity has been demonstrated in the hyperplastic membrane of the thickened diaphysis and subperiosteal proliferation of magnesium-deficient rats (Bélanger et al., 1972), which also showed the more typical epiphyseal growth suppression. This observation supports the premise that the high level of the enzyme lowers that of the inhibiting polyphosphates, allowing for increased mineralization of the diaphysis. Why this magnesium-dependent enzyme should be found in such high concentrations in the membrane of the bone shaft of magnesium-deficient animals requires resolution. Similarly, more study is needed into why the increase in bone shaft alkaline phosphatase of magnesium deficiency should be associated with hyperplasia, resembling desmoid tumors, that was characterized by more fibrous tissue in parathyroidectomized animals, more bone formation when PTH was given, and less subperiosteal hyperplasia when estradiol (an alkaline phosphatase stimulator: Malinow et al., 1960) was given. Another puzzling observation is the association of osteogenic sarcomas with beryllium, which inactivates alkaline phosphatase, possibly replacing the activating magnesium (Grier et al., 1949; Aldridge, 1950).
The bits of evidence that patients with genetic bone dysplasia have abnormal (usually low) bone phosphatase levels, and that low magnesium levels lead to abnormal matrix formation and to defective osteocytic differentiation, suggest that normal magnesium utilization might be at fault. Evaluation of the magnesium status and bone phosphatase levels and activity of patients with genetic or neoplastic bone disease, and of the effect of magnesium on the enzyme activity of the biopsies, might prove worthwhile. If it would lead to prophylactic or therapeutic approaches remains to be seen.
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
{kind=link}
{kind=link}
{kind=link}