MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
*NO figures and tables for Chapter 8*
Part II: Chapter 8
MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF CARDIOVASCULAR DISEASES
8
Clinical Cardiac Abnormalities and Magnesium
There are several clinical conditions associated with cardiac abnormalities that resemble those produced by experimental magnesium deficiency or that cause loss of myocardial magnesium. Bajusz (1965b) refers to experimental necrotizing cardiomyopathies to describe a variety of degenerative processes that are more or less confined to the myocardium. He commented that the disease is characterized by subendocardial necrotizing foci, usually without significant disease of the coronary vessels, although comparable disorders can be produced by thrombogenic diets. He observed that "coronary heart diseases" should be classified as primary or secondary cardiomyopathies that result from vascular factors (e.g., coronary artery spasms, local microcirculatory changes), factors that directly affect myocardial metabolism and the susceptibility or resistance of the myocardium at the time of potentially cardiotoxic episodes. He pointed out that, in addition to stress situations, hormones, and age, cardiomyopathy-conditioning factors include sodium excess and deficiencies of chloride, and especially of potassium and magnesium. Bajusz (1969) has stated that the loss of these two cations from the myocardial cells (that are associated with early ultrastructural changes such as enlargement and vacuolization of the sarcoplasmic reticulum and mitochondrial degeneration) seem to be important components of many, if not all types of disturbances in cardiac metabolism, resulting in myocardial degeneration, heart failure, or fatal conduction defects.
Cardiomyopathies (and dysrhythmias not caused by diagnosed ischemic heart disease) are generally classified by clinical manifestations or by pathologic characteristics. In a criticism of any classification of primary (or secondary) cardiomyopathies that is dependent on postmortem diagnosis, Mattingly (1970) suggested that what is needed is greater effort directed toward recognition of early clinical features, search for etiologic factors, study of biochemical as well as hemodynamic alterations, and search for iatrogenic factors in the pathogenesis of and for control of this disease. Because magnesium deficiency or loss can be correlated with many of the cardiomyopathies for which a cause has been established, perhaps it participates in those considered primary or idiopathic.
8.1. Cardiomyopathies Not Secondary to Disease of the Major Coronary Arteries or to Infection
The term "cardiomyopathy" was introduced by Brigden (1957) to indicate isolated noncoronary myocardial disease, present without significant disease of other systems of the body. He included instances of familial disease, amyloidosis, alcoholism, a few postpartum, and several that seemed to be due to infection. Restriction of the term to isolated myocardial disease might be unwise, since it excludes very similar cardiomyopathies associated with diseases that might provide clues to a common pathogenic factor. In subsequent surveys, the term has been used as synonymous with "myocardial disease," to indicate dysfunction of the muscular pump that is not the result of structural deformity of the heart, hypertension, or coronary atherosclerosis (Editorial, Brit Med J, 1969). Goodwin (1970) classifies them as congestive and hypertrophic. Hudson (1970) suggests four features that characterize cardiomyopathies: cardiomegaly, endocardial thickening, mural endocardial thrombus, and myocardial scars. He considers the fibrotic cardiomyopathies (sometimes accompanied by foci of myocardial calcification) to be likely to be related to hyperreactivity to vitamin D in infancy. He cites myocardial fibrosis in adults as the commonest form of idiopathic cardiomyopathy; it can be familial or occur as isolated cases. Hypertrophic obstructive cardiomyopathy is also familial in some instances, and Hudson (1970) considers it congenital. It is associated with narrowed outflow from either or both ventricles. Among the list of conditions that Hudson (1970) considers possibly contributory to cardiomyopathy are several in which magnesium deficiency or loss have been described: the peripartal state, infantile fibrotic cardiomyopathy, or adult endocardial fibrosis, myofibrosis (or a combination of both), alcoholism, calcium oxalosis, calcific degenerative processes, hyperparathyroidism with myocardial calcification, protein calorie malnutrition, beriberi (Oriental, or the similar, but vitamin-B1-resistant forms seen in alcoholism and in the peripartal period), anemia, severe diarrhea, toxicity from catecholamine or cardiac glycosides, and severe trauma.
Cardiomyopathies occur throughout the world (Shaper, 1968; Editorial, Brit Med J, 1969). In the symposium on "Experimental 'Metabolic' Cardiopathies and Their Relationship to Human Heart Disease" (Ann NY Acad Sci 156, 1969), J. N. Davies (1969) discussed the African cardiomyopathies with and without endocardial lesions, and commented that none of them are confined to the African continent, as is suggested by the name. He suggested that, because coronary arteriosclerosis is so common in the United States, it is possible that many cases of cardiomyopathy are missed. This recalls Caddell's (1965, 1969b) comment that the endomyocardial fibrotic disease in Africa is prevalent in areas where protein calorie malnutrition (associated with magnesium deficiency in the recovery syndrome) is found, and her suggestion that prolonged electrolyte imbalance might be contributory.
T. James (1967) proposed that disease of the small coronary arteries might be an important contributory factor in cardiomyopathies of obscure origin, a position taken also by Varnauskas (1967). James (1967) points out that the clinical manifestations of cardiomyopathy (progressive cardiac enlargement and failure without clear cause, an inordinately high incidence of arrhythmias and conduction disturbances, syncope, sudden unexpected death, and atypical chest pain) can all be due to abnormalities of the small coronary arteries. Considering a basic abnormality to be disease of the cardiac microcirculation permits inclusion of diabetic cardiomyopathy and the cardiomyopathy of progressive muscular dystrophy (James, 1962) in the same category. Neonatal coronary arteriosclerosis-cardiomyopathy complex (Seelig and Haddy, 1976/1980) can be similarly categorized. It further substantiates the premise that loss of myocardial magnesium (and potassium) is likely to be common, not only to experimental but to clinical cardiodegenerative processes (Bajusz, 1965a; Lehr et al., 1966; Lehr, 1969; Lehr et al., 1976/1980), abnormalities of the myocardial microcirculation having been implicated in several of the experimental models that cause both microfocal myocardial necrosis and magnesium loss (Lehr, 1964, 1965, 1966, 1969).
There is no direct evidence that isolated or familial "idiopathic" cardiomyopathy is caused by magnesium loss or deficiency. Ultramicroscopic examination has shown some similarities to those seen in experimental magnesium deficiency and to some of the diseases in which magnesium loss has been described. For example, Hudson (1970) has examined myocardium from hypertrophic cardiomyopathy and shown sarcomeres with widely separated Z bands. Bulloch et al. (1972) found that myocardial biopsy specimens from 12 patients with idiopathic cardiomyopathy were similar to that associated with alcoholism.
8.1.1. Peripartum Cardiomyopathy
The etiology of peripartum heart failure is still a mystery (Editorial, Brit Med J, 1976a), even though it has long been recognized. It was reported in an 1848 textbook by Meigs, and myocardial degeneration was described in women who died in the peurperium by Virchow in 1870. It was reported sporadically in the first third of this century as an important factor in producing heart failure in the peripartal period (Review: Gouley et al., 1937), and is now accepted as definite entity of unknown etiology that is listed as a cause of "primary" cardiomyopathy (Brigden, 1957; Hudson, 1970). The hemodynamic load of pregnancy has been implicated, but in editorial evaluations of this problem, nutritional inadequacy to meet the demands of pregnancy and lactation were considered more likely (Editorials, Brit Med J, 1968, 1976a).
Although magnesium deficiency has not been considered a possible nutritional factor in peripartum cardiomyopathy, there is considerable circumstantial evidence that points to magnesium depletion. Among the conditions associated with peripartal cardiac failure are those that predispose to preeclampsia and eclampsia, with which it is often associated-maternal immaturity, multiple births, and high parity-especially when rapidly successive (Hull and Hafkesbring, 1937; Hull and Hidden, 1938; Teel et al., 1937, Melvin, 1947; Szekely and Snaith, 1947; Walsh et al, 1965; Govan, 1966; Stuart, 1968; J. B. Johnson et al., 1966). These are all conditions that predispose to maternal magnesium depletion. Furthermore the cardiac lesions of peripartum cardiomyopathy (supra vide) strikingly resemble those of experimental "pure" magnesium deficiency
Melvin (1947) and earlier Gouley et al. (1937) and Hull and his colleagues (1937, 1938) commented on the similarity of postpartum heart disease to cardiac beriberi, and implicated probable nutritional inadequacy. However, despite the similarity of manifestations of the disease to Oriental beriberi cardiomyopathy, it is refractory to thiamine therapy (Melvin, 1947; Stuart, 1968). This recalls the refractoriness of alcoholic "beriberi" cardiomyopathy to thiamine and to the dependence of vitamin B1 on magnesium (p. 215). The relatively high frequency of puerperal and "idiopathic" cardiomyopathy in Jamaica, usually in patients with histories of poor nutrition (Walsh et al., 1965; Stuart, 1968), recalls the early demonstration of bovine cardiovascular lesions in Jamaica that were deemed likely to be caused by a "conditioned" magnesium deficiency (Arnold and Fincham, 1950).
Review of the literature shows that the clinical picture is usually one of heart failure, presenting with shortness of breath, palpitations, edema (rarely with acute pulmonary edema), precordial pain, and embolism (Gouley et al., 1937; Teel et al., 1937; Hull et al., 1937; 1938; Szekely and Snaith, 1947; Brigden, 1957; Meadows, 1957; S. Rosen, 1959; Benchimol et al., 1959; Seftel and Susser, 1961; Gilchrist 1963; Walsh et al., 1965; J. B. Johnson et al., 1966; Stuart, 1968; Demakis and Rahimtoola, 1971). Diastolic, and less frequently systolic, hypertension are often found, as is cardiomegaly and abnormal ECGs. Stuart (1968) has commented that close cardiac surveillance may disclose symptomless cardiomegaly or abnormal ECG in an apparently well woman. Toxemia is commonly, but not invariably, part of the history of women who develop peripartal heart failure; Govan (1968) found that cardiorespiratory failure was the cause of fatal eclampsia in his series of 110 cases.
Hypertrophic obstructive cardiomyopathy has been reported by G. Tuner et al. (1968) as an increasingly recognized and often familial form of cardiomyopathy of pregnancy. The Editorial (Brit Med J, 1968) that called attention to this now more common form of cardiac disease of pregnancy suggested that some of the young pregnant women with angina and tachycardia, with ECG abnormalities that persisted after pregnancy (Gilchrist, 1963), might have had this abnormality. This possibility calls to mind the epidemic of supravalvular aortic stenosis syndrome, and other outflow obstructive lesions, that were associated with hyperreactivity to vitamin D at the time of excessive fortification of milk with vitamin D or its use in massive parenteral dosage (Review: Seelig, 1969b) especially in the late 1940s through the 1950s. Is it possible that some of the infants so treated might have been insufficiently hyperreactive to vitamin D to develop the full-blown syndrome, but might have developed silent outflow-obstructive lesions that became overt during the peripartum period? Possibly the presumptively vitamin-D-hyperreactive women might also have had myocardial lesions as a result of the vitamin-D-induced loss of magnesium during infancy and might have been unduly susceptible to both vitamin D and magnesium deficiency during pregnancy
There are several additional fragments of circumstantial evidence suggestive of magnesium deficiency.
1. Patients with this disease have been found to be unusually susceptible to digitalis toxicity, developing multiple premature ventricular contractions that sometimes persist (Walsh et al., 1965; Demakis and Rahimtoola, 1971). (The susceptibility of magnesium-deficient dogs and monkeys to digitalis toxicity should be recalled here.) J. B. Johnson et al. (1966) reported an ultimately fatal case of a 14-year-old mother of twins who, because of her age and the twin pregnancy, had almost certainly been deficient in magnesium. She was unduly sensitive to digitalis.
2. In toxemia, there is commonly aldosteronism and sodium retention (A. Barnes and Quilligan, 1956), and increased catecholamine secretion (Zuspan, 1972), hormones that are secreted in excess in magnesium deficiency and that cause magnesium loss. In addition, women with preeclamptic or eclamptic pregnancies have an exaggerated response to catecholamine infusions (Raab et al., 1956; Zuspan et al., 1964), which have been used as a prognostic test in preeclampsia (Raab, 1957) and to differentiate between essential hypertension and toxemia of pregnancy (Zuspan et al., 1964). The combination of excessive "stress" hormones with probable magnesium deficiency puts cardiomyopathy of pregnancy squarely into the category of "pluricausal" dysionic cardiomyopathy.
3. The susceptibility to peripartal intravascular coagulation, with the risks of damage to the placenta (Bonnar et al., 1971) and of maternal death from embolic phenomena (Arthure, 1968) or that have been difficult to control even with anticoagu1ants (S. Rosen, 1959), might also be related to magnesium deficiency.
4. Among all of the cases reviewed, there was mention of use of magnesium in only two instances: one to measure circulation time, and the other in the management of eclampsia. Decherd and Herrmann (1944) commented briefly that severe tachycardia (of a woman who had had toxemia of pregnancy and developed postpartum heart failure) disappeared after diagnostic intravenous injection of magnesium sulfate. The arrhythmia later recurred and was treated traditionally. In the other instance, magnesium therapy (presumably high dosage) was given to a woman who developed eclampsia and cardiac decompensation during her fourth pregnancy (Teel et al., 1937). The authors noted her "rapid recovery" and lack of recurrence of cardiac manifestations even when she returned, again pregnant, several years later. This was in contrast to five other patients with peripartal cardiomyopathy in this series: Two died and the others required digitalization, two for a short period, one of whom had persistent ankle edema and one of whom had a protracted and incomplete recovery. Whether the magnesium therapy played any role in the complete, rapid recovery of the patient remains speculative. A 38-year-old patient in this series, who died suddenly after she developed cardiac asthma and anasarca during the seventh month of her eighth pregnancy, had myocardial edema, but not necrosis, and slight subendocardial necrosis. She apparently died early in the course of the disease (possibly of arrhythmia), and thus the changes of the small myocardial arteries (intimal hyperplasia and elastica thickening) are of particular interest, since they resemble the changes of experimental magnesium deficiency.
Necropsy examination of patients who died of peripartum cardiomyopathy generally discloses cardiomegaly and dilatation, with focal or diffuse myocardial necrosis and (in later instances) fibrosis, endocardial edema, necrosis and fibrosis and mural thrombi (Gouley et al., 1937; Meadows, 1957; Walsh et al., 1965; J. Johnson et al., 1966; Ledingham et al., 1968; Hudson, 1970; Sakakibara et al., 1970). Several pathologists have reported thickened myocardial arterioles, some times with intimal edema or hyperplasia (Gouley et al. 1937; Teel et al., 1937) and perivascular infiltration around the small coronaries (Meadows, 1957).
Biopsy specimens were examined ultramicroscopically in two reported instances. Perinuclear hydropic vacuolization of myocardial fibers and sarcoplasmic fragmentation was seen 2 months before death from progressive heart failure [7 months after a twin delivery by a 14-year-old girl (J. B. Johnson et al., 1966)]. A 30-year-old woman, who survived the cardiomyopathy that became manifest a week after delivery of her second baby, had widened sarcoplasmic spaces containing irregularly shaped electron-dense deposits, as well as vacuolization (Sakakibara et al., 1970).
8.1.2. Infantile Cardiomyopathy
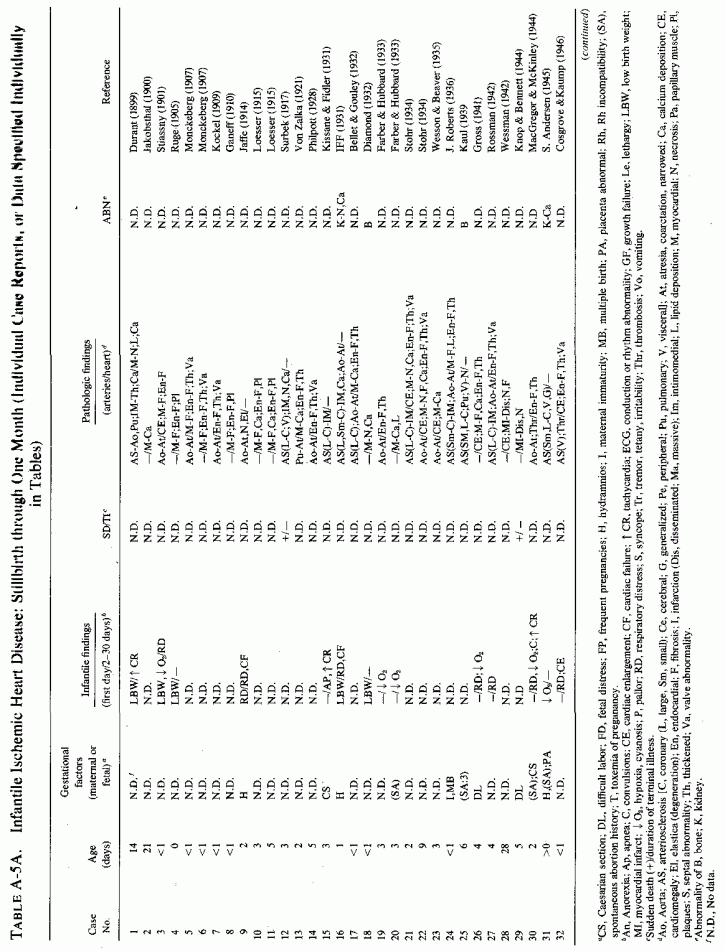
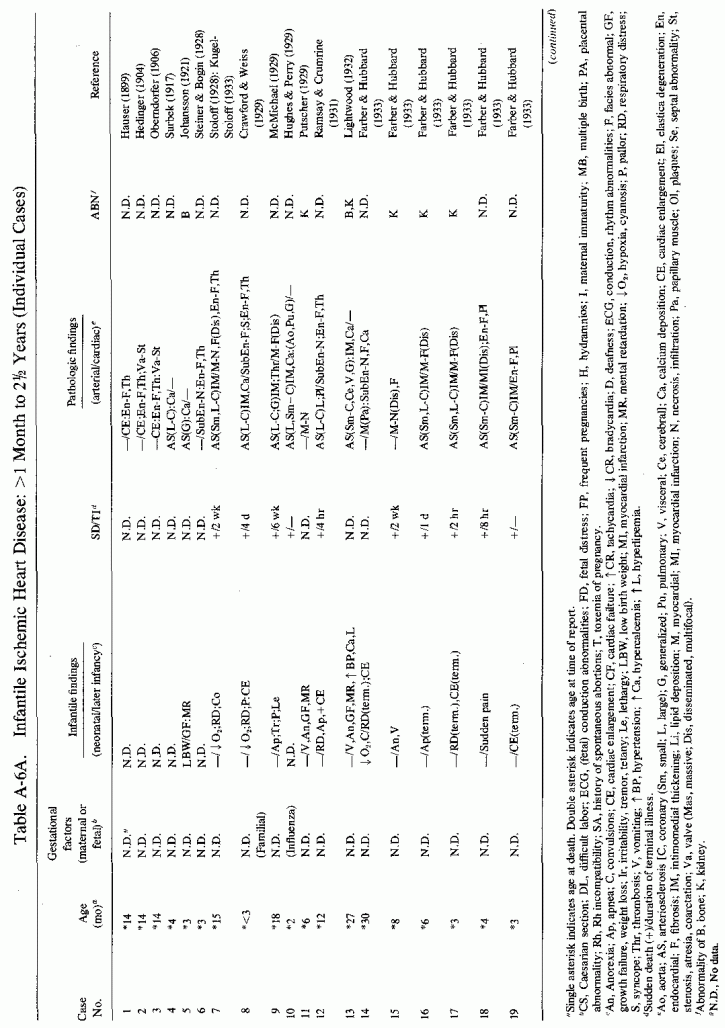
Coronary and generalized arteriosclerosis of infancy has received more attention in the literature than has infantile cardiac disease (if one excludes the valvular abnormalities and the great vessel and peripheral pulmonary atresias). However, many reporting infantile cardiovascular lesions also mention myocardial and endocardial lesions. Among the lesions tabulated (see Appendix Tables A-5A, A-5B and A-6A, A-6B89.) alone or in combination, are multifocal myocardial necrosis (such as is seen with the small coronary artery damage of magnesium deficiency, subendocardial and papillary muscle necrosis and fibrosis, and endocardial fibroelastosis, as well as massive myocardial infarctions. Among the 157 individual case reports of infants who were stillborn or who died in the first month of life, 37 had myocardial necrosis or cellular infiltration, 23 had myocardial calcinosis, and 38 had myocardial fibrosis. Among the individually cited 253 infants between 1 month and 2 ½ years of age, 72 had necrotic myocardial lesions, 19 had calcific lesions, and 42 had myocardial fibrosis. Endocardial fibroelastosis was reported in 83 of the infants under 1 month of age and in one-third of those of 1 month to 2 ½ years. Over half of the younger group of infants with EFE had outflow obstruction; only about a quarter of those between 1 month and 2 1/2 years of age, tabulated individually, had outflow obstruction. This is in contrast to the surveys of patients selected for EFE, among whom outflow obstruction was found to be very common (Moller et al., 1964; J. Edwards et al., 1965; Oppenheimer and Esterly, 1966). Perhaps a reason for the contrasting findings is the age limitation in cases tabulated and reviewed. Congenital outflow abnormalities-whether the supravalvular aortic stenosis syndrome (SASS), aortic or pulmonic atresia or peripheral pulmonary artery stenoses (alone or in combination)-are also commonly associated with coronary, endocardial, or myocardial diseases and with hyperreactivity to vitamin D (Beuren et al., 1964, 1966; Peterson et al., 1965; Taussig, 1966). Subvalvular aortic stenosis has also recently been suggested as a possible result of hypervitaminosis D (McFarland et al., 1978). There is a relatively small representation of children with outflow abnormalities and cardiofacial peculiarities (which have received much recent attention as familial and isolated cases) in the Appendix tables limited to infants up to 2 ½ years of age. When the endocardial thickening or the arterial disease involves the septum and conducting tissue, arrhythmias and cardiac arrest might result in chronic cardiac disease or in early death. The conditions seen in those surviving beyond infancy include arrhythmias and syncopes. The implication of hypervitaminosis D in such conditions, and the description of calcification of the labyrinth in infants with outflow obstruction, with and without endocardial fibroelastosis and cardiofacies (see cited publications by Beuren et al., 1962, 1964, 1966, in Appendix Table A-6B) raises the question as to whether the syndrome of deaf-mutism, prolongation of the Q-T interval, syncope, and sudden death in children and young adults (Jervell and Lange-Nielsen, 1957) (see cases 187-189, 193, 194, 203, 204, 233: Appendix Table A-6A) might be disorders in which susceptibility to vitamin D toxicity or magnesium loss or malabsorption might play an etiologic role.
In the young infants, prodromal symptoms preceded death by only a few hours to a few days. The symptoms presented are not unlike those reported for infants who died of SIDS. Some of the babies with cardiovascular abnormalities, proved at autopsy, had had signs of illness from the time of birth. ECG tracings typical of ischemic heart disease, were sometimes obtained. Those who had a subacute or chronic course generally were flaccid and quiet, behavior similar to that described by Naeye (1976a) in the SIDS. Those who did not die suddenly or after a short illness of sudden onset generally had had a fairly steady downhill course, with sustained anorexia, vomiting, weight loss, and debility. Several developed hypertension. Coronary arteriosclerosis and focal myocardial necrosis and fibrosis have been found in infants who died suddenly and in others who had been ill with clinically manifest heart disease, many of whose first cardiac manifestations developed at about two to four months of age, the age of peak incidence of SIDS. An international study of 254 cases of sudden unexpected death from cardiovascular disease (in which infants under a year of age were excluded to eliminate the SIDS) found that those who died from 1 to 5 years of age had a disproportionate representation of EFE, pulmonary stenosis, and A-V block (Lambert et al., 1974). Almost a tenth of the total cases were familial. The sudden deaths of the entire series of deaths from 1 to 21 years were associated with myocardial hypoxia in half; about a third had arrhythmias.
Similar total cardiomyopathies, developing postpartum in a mother and in her 7-year-old daughter (Hudson, 1970), raises the possibility that this may have been an instance in which gestational malnutrition (magnesium?) deficiency might have caused maternal and fetal cardiac damage.
The neonatal hypoparathyroidism and hypomagnesemia of infants fed cows' milk might be the human counterpart of the model of cardiorenal necrosis, produced by sodium phosphate loading of parathyroidectomized rats (Lehr et al., 1966; Lehr, 1969). Such infants have a high phosphate/magnesium ratio. Since excesses of both calcium and phosphate (relative to magnesium) are cardiopathic, the prevalence of dietary customs that lead to such imbalances perinatally and in early infancy might be contributory to cardiomyopathy of infants and young children. Persistence of such nutritional imbalances, which might become worse as the intake of high phosphate sodas increases, and as alcohol ingestion begins, can intensify cardiomyopathic lesions that, like the arterial lesions that receive more attention, might have their roots in infancy and possibly even before birth. Since magnesium deficiency causes damage to the intramural small coronary arteries, the perivascular damage to the myocardium that has been reported is not surprising. As in the experimental model, infants who died of cardiovascular disease typically have microfocal myocardial necrosis, infiltration, and fibrosis.
Myocardial mitochondrial and cytoplasmic changes have also been reported. Mitochondria obtained by needle biopsy of a 6-month-old boy with respiratory distress and congestive heart failure had closely stacked, parallel, concentrically arranged cristae, with some cristae filled with electron-dense granular material (Hug and Schubert, 1970). These characteristics are similar to those reported in magnesium-deficient rats (Heggtveit et al., 1964; Heggtveit, 1965b,c). They were not found in the myocardium (at autopsy) of a 6-year-old girl with idiopathic cardiomyopathy, in which there was dissolution of the myofibrillar structures (Hug and Schubert, 1970). Lin (1972) described extensive mitochondrial calcification in the myocardium of a 10-week-old baby boy, who had postductal coarctation, and had had several episodes of cardiac arrest lasting 10 to 50 minutes. The intramitochondrial deposits were needle-shaped dense crystals that resemble those described by Silver and Sordahl (1976/1980) in their in vitro studies of cardiac mitochondria in magnesium-free medium. Lin (1972) noted that ischemia produces intramitochondrial dense bodies that probably represent calcium accumulation, and that the magnesium and potassium contents of ischemia-damaged mitochondria were reduced (Jennings, 1969). An autopsy was obtained 5 hours after the death of a 16-month-old girl who had been in good health until sudden onset of pallor and rapid pulse, with supraventricular tachycardia (320/minute), 18 days antemortem (Haese et al., 1972). The heart showed numerous swollen rounded myocardial cells with partial or complete loss of contractile elements and granular or vacuolated sarcoplasm. Occasional necrotic myocardial cells had adjacent inflammatory cells. The altered cells had many lipid droplets. The mitochondria were distorted. Similar myocardial lesions had been reported in four other female infants (Ross and Belton, 1968; J. Reid et al., 1968; MacMahon, 1971). Of the 13- and 16-month-old baby girls reported by Reid et al. (1968), the first died suddenly while playing, with no prior evidence of illness. The second was admitted with a history of vomiting, drowsiness, and left hemiplegia after a fall. She was found to have right bundle branch block and supraventricular tachycardia. She was unresponsive to therapy, developed new thrombotic events, and died 3 days after admission. Reid et al. (1968) considered the abnormal cells in the myocardium and in the region of the atrioventricular node as a probable reaction to degenerating myocardial fibers. The 13-month-old girl reported by MacMahon (1971) was the seventh child; the preceding sibling had had multiple developmental anomalies, including cardiac disease, and died at 16 months of age. The propositus had been well until 15 hours before admission. Repeated episodes of vomiting and then tachycardia led to hospitalization; ECU showed arrhythmia and a rate of 200/minute. Half an hour after digitalization and starting intravenous fluids, ventricular fibrillation developed. Recurrent episodes were treated by external cardiac massage, defibrillation, and finally adrenalin, calcium chloride, and isoproterenol. The next day she developed tonic-clonic seizures. She died 62 hours after admission, and at autopsy had many "xanthoma cells" throughout the myocardium, in the subendocardium and in the septum, involving the conducting system. No data were given as to the intervals between the births of the patient and her six siblings, but the multiple anomalies of the immediately preceding baby suggest that the mother might have been nutritionally depleted, possibly of magnesium. Thus, her last infant might also have been low in magnesium stores, and might have had small coronary arterial disease such as has been implicated in conduction tissue disease.
Another baby girl (8½ months old) first developed an episode of paroxysmal atrial tachycardia (PAT) that responded to digitalis about 2 months before her death Bove and Schwartz, 1973). The PAT recurred 3 days before her death (while she was still on digitalis), and she was treated with direct current shock and pacing, to which she was unresponsive, developing profound hypotension necessitating administration of epinephrine and isoproterenol. Necropsy examination showed microfoci of acute ischemic necrosis and cells resembling storage histiocytes, containing lipid, scattered throughout the left ventricular wall, the interventricular septum, and both atria. Ultramicroscopy showed mitochondria, many of which were swollen and contained amorphous dense inclusions. In focal areas the cristae were stacked; the outer membranes of adjacent mitochondria were fused to form electron-dense segments. There were focal aggregates of swollen lipid-laden myocardial fibers and myofibrillar membrane-limited dense granular that seemed to be spatially related to early Z-band degeneration. These findings resemble those described under magnesium deficiency. Possibly, the lipid accumulation in this and the preceding case might have been contributed to by the catecholamines given in an effort to correct the hypotension. It is conceivable that the refractory hypotension of these infants might have been the result of magnesium depletion; in magnesium deficiency, in vitro, arterial smooth muscle exhibits markedly diminished arterial contraction in response to vasoactive amines (pages 179-183).
8.1.3. Alcoholic Cardiomyopathy and Magnesium Deficiency
Alcoholic cardiomyopathy has been considered a nutritional disease, caused predominantly by thiamine deficiency and by deficiencies of other vitamins (Blankenhorn, 1945). There was then a shift in emphasis, implicating a directly cardiotoxic effect of alcohol, since thiamine is not therapeutic in a substantial number of chronic consumers of hard liquor, and many of the patients are well nourished and respond to prolonged bed rest and abstinence from alcohol (Burch and DePasquale, 1969). More recent work focuses attention on the nutritional aspect of the disease, but this time with the major emphasis on magnesium deficiency as a common denominator in the failure to respond to thiamine, in the arrhythmias seen in alcoholic cardiomyopathy, and in the cardiac lipid accumulation and ultramicroscopic changes.
Thiamine loses enzymatic activity in magnesium-deficient rats, which exhibit signs of thiamine deficiency unless magnesium is repleted (Zieve et al., 1968a,b; Zieve, 1969). Furthermore, thiamine levels have been shown to fall in liver and kidneys of magnesium-deficient rats (Itokawa et al., 1974c). Magnesium-deficient alcoholics are unresponsive to vitamin B (and other B vitamins) until their magnesium is repleted (Zieve, 1975). Magnesium deficiency has long been recognized in alcoholism (Flink et al., 1954; Review: Rink, 1976/1980); it can be secondary to low intake, malabsorption, and, if cirrhosis develops, secondary aldosteronism (Review: Massry and Coburn, 1973). The dependence of thiamine activity on magnesium as a co-factor is relevant (not only to the psychoneurologic manifestations of alcohol withdrawal) but to at least three of the metabolic aberrations that affect the heart in alcoholism.
1. Itokawa et al. (1973) have found that there is increased lipogenesis, both in magnesium-deficient and in thiamine-deficient rats. They demonstrated increased lipid and cholesterol in liver and kidneys and hypothesize that these deficiencies lead to a general increase in lipid synthesis, possibly by blocking the pathway of acetate to the tricarboxylic cycle, shunting the acetate to the lipogenesis pathway. Thus, it is possible that the magnesium-thiamine deficiencies are contributory to the accumulation of myocardial lipid droplets that have long been recognized as characteristic of alcoholic cardiomyopathy. Thiamine deficiency has also been shown to cause myocardial catecholamine accumulation (Raab and Supplee, 1944), an effect also demonstrated for magnesium deficiency.
2. Another metabolic aberration to which magnesium-thiamine deficiency might contribute is acetaldehyde accumulation, which might result from blockage of the thiamine-dependent step by which acetaldehyde goes to pyruvate (Altman and Dittmer, 1968). Discussed elsewhere in this volume is the acetaldehyde-induced arrhythmia (which has been shown to be protected against by β-adrenergic blockade), suggesting that it is mediated by catecholamine release, but that might just as well be mediated, in alcoholism, by magnesium deficiency. Contributory to the presumed increased catecholamine effect might be ethanol's interference with catecholamine metabolism (V. Davis et al., 1967b).
3. There is an interrelationship between magnesium deficiency and thiamine (excess) that bears on serotonin levels and metabolism (Itokawa et al., 1972b). Magnesium (as the EGTA chelate) inhibits the release of serotonin from platelets (Henson, 1969). Magnesium deficiency, particularly in the presence of excess thiamine, inhibits the oxidation of serotonin (Itokawa et al., 1974a). Whether ethanol's interference with serotonin's metabolism (V. Davis et al., 1967a) might be intensified by magnesium deficiency and thiamine therapy might be worth investigating.
Further inferential evidence that magnesium deficiency is contributory to alcoholic cardiomyopathy derives from study of the cardiac lesions. (Heggtveit 1965a), who had observed that the coronary arterioles of magnesium-deficient rats were edematous, also observed that intravenous infusion of 20% ethanol into rats caused significant swelling of the capillary endothelial cells (Heggtveit and Nadkarni, 1971).
Pintar et al. (1965) reported edema and disorganization of the layers of the coronary arterioles, with perivascular foci of edema, necrosis, and spotty calcification in the hearts of three alcoholic men, 53 to 63 years of age. One also had subendocardial fibrosis. Their aortas and major branches of their coronary arteries showed only minimal atheromatous changes. Alcoholics who died with early stages of cardiomyopathy were reported to have edema of the coronary vessels (Benchimol and Schlesinger, 1953). Three patients with advanced alcoholic cardiomyopathy, who had died after developing clear signs of acute transmural infarction, had a coronary obstruction but had periarterial myocardial fibrosis (Regan et al., 1975). These investigators speculated that the fibrosis around the myocardial coronary arteries might have interfered with their ability to dilate, thereby causing confluent necrosis when oxygen requirements increased.
Pintar et al. (1965) suggested that the vessel wall edema of their alcoholic cardiomyopathic patients might have resulted from hypomagnesemia. Recently, Hungerford and Bernick (1976/ 1980) gave histologic details of the coronary arterial structural disorganization caused by magnesium deficiency.
Ultramicroscopic studies of alcoholic cardiomyopathic hearts and hearts from magnesium-deficient animals also show similarities. Heggtveit and Nadkarni (1971) reported significant swelling of the mitochondria and sarcoplasmic reticulum after an acute ethanol load in rats, but were unable to induce cardiomyopathy by long term alcohol feeding. Szanto et al. (1967) however, did find some changes in the mitochondria and sarcoplasmic reticulum of alcohol-fed rats. Mice seem to be more susceptible to alcohol cardiomyopathy. Sohal and Burch (1969) found strikingly separated intercalated discs of mice given water containing 15% ethanol for three weeks. Burch et al. (1971) then reported that alcohol (ethanol, beer, or wine) produced myocardial damage in mice even when they are otherwise properly fed. The mice developed swelling of the mitochondrial cristae and of the sarcoplasmic reticulum, disorientation of the myofibrils, expansion of the intercalated disc, and accumulation of fatty deposits and dense particles. These changes are very like those reported by Hibbs et al. (1965) in six autopsied cases of alcoholic cardiomyopathy: severe mitochondrial swelling, degeneration and fragmentation of the cristae, and formation of dense inclusions. There was also pronounced swelling of the sarcoplasmic reticulum, excessive lipid accumulation, and myofibrillar degeneration and lysis.
Needle biopsy specimens from patients with alcoholic cardiomyopathy also showed severe mitochondrial changes, with subsequent derangement and fragmentation of contractile elements (Alexander, 1966b). This investigator commented that the ultramicroscopic changes resemble those of magnesium-deficient animals, and suggested that the myocardial lesions might be secondary to ethanol-induced magnesium deficiency, rather than a direct consequence of the alcohol per se. He referred to the evidence that the metabolism of the vasoactive amines, catecholamine and serotonin, is interfered with by ethanol, and considered the possibility that their accumulation in cardiovascular tissue of alcoholics might thereby be enhanced.
Myocardial biopsy specimens from eight patients with alcoholic cardiomyopathy, and from twelve with idiopathic cardiomyopathy were compared by Bulloch et al. (1972). The major ultrastructural lesion in both was contractile element-sarcoplasmic reticulum disorganization. Swelling of the sarcoplasmic reticulum was early and generalized in the alcoholic cardiac disease; it was focal and inconstant in idiopathic cardiomyopathy. In this series of cases, mitochondrial damage was not a major lesion in either diseases.
The similarity of the ultramicroscopic findings in these two diseases of such diverse etiologies, and their similarities to the changes of magnesium deficiency and of a variety of experimental models characterized by loss of myocardial magnesium suggest that testing for tissue losses of magnesium, and trial of magnesium supplementation be investigated.
8.1.4. Diabetic Cardiomyopathy
Diabetes mellitus is one of the diseases that was first recognized to be associated with magnesium deficiency (Martin and Wertman 1947; Martin et al., 1951 1958; Martin, 1969, Jackson and Meier, 1968). Additionally, diabetics commonly have diffuse endarterial proliferative small vessel disease (Ditzel, 1954) that resembles the arteriolar lesions of magnesium deficiency. In a study of small vessel disease of the diabetic heart, Rubler et al., (1972) presented cases with fibrosis throughout the myocardium, in conjunction with the damaged intramural vessels Among 73 patients with "idiopathic" primary cardiac disease, studied by Hamby et al. (1974) 16 had diabetes mellitus, a high frequency of diabetes that was statistically significant. Autopsies were performed in 3 of the 4 diabetic patients with cardiomyopathy who died; all 3 had small coronary, but not large coronary artery disease. In this series of cases, only one of 28 patients with cardiomyopathy without diabetes mellitus had small coronary vessel disease.
T. James (1967) suggested that arrhythmias of diabetes mellitus might be caused by small coronary arterial disease of the vessels supplying the conducting tissue of the heart. Impaired atrioventricular conduction has been found significantly more frequently in patients with diabetes mellitus or abnormal glucose levels (Rubler et al., 1975) than in patients with other diseases.
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
*NO figures and tables for Chapter 8*
{kind=link}
{kind=link}
{kind=link}
{kind=link}