MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
Part I: Chapter 3
MAGNESIUM DEFICIENCY DURING GESTATION, INFANCY, AND EARLY CHILDHOOD
3
Consideration of Magnesium Deficiency in Perinatal Hormonal and Mineral
Imbalances
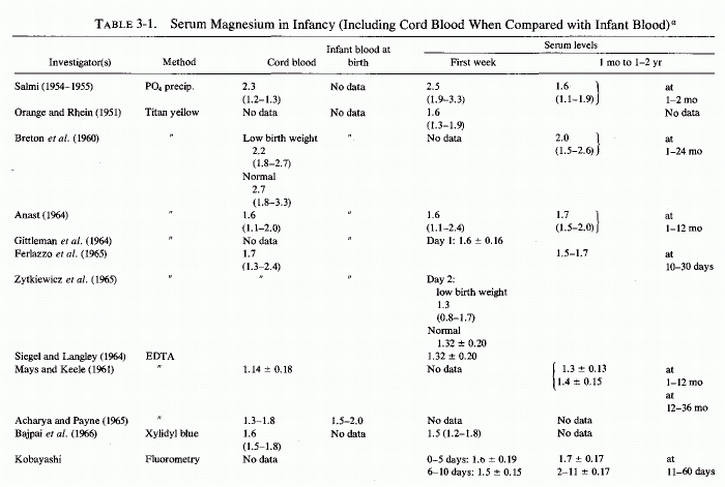
In view of the evidence that inadequate magnesium intake is common during pregnancy and that the plasma levels of magnesium tend to fall, especially during the first and third trimesters of pregnancy even when corrected for hemodilution, it is not surprising that neonatal magnesium deficiency can create problems. Until relatively recent years, however, measurement of magnesium levels in infants was rare. Cord blood analyses, done at intervals since 1923 (Table 3-1) and (Table 3-1 continued) showed wide ranges reported in individual studies, even when the quite reliable old precipitation methods or the more reliable modem procedures were employed. Since individual maternal status and infant status were not designated in most instances, these wide ranges are difficult to interpret. Low levels may have reflected maternal and fetal insufficiency; high levels may have reflected magnesium therapy for preeclampsia. Mean values are even more difficult to evaluate. Determination of serum or plasma magnesium levels of the infant at birth or within hours thereafter presents more problems. Intrauterine asphyxia, difficulties in delivery, or other causes of birth hypoxia or acidosis, and hyperosmolality can all contribute to elevations of serum magnesium levels as the cellular magnesium is released to the extracellular fluid, changes similar to those seen with surgical and other traumatic shock and hypoxic conditions. Such infants have been found to have a negative correlation between their serum magnesium levels and their Apgar scores (Engel and Elm, 1970; Jukarainen, 1974). Infants who are hypermagnesemic when born shortly after their eclamptic mothers had received pharmacologic parenteral doses of magnesium also are likely to be depressed and have low Apgar scores. The first group of infants is likely to be cellularly depleted of magnesium, which becomes manifest as hypomagnesemia, usually by the fifth day of life. Those with hypermagnesemia following maternal magnesium therapy usually take longer for their serum levels to drop to normal limits. If the infant survives the respiratory depression of pharmacologic hypermagnesemia, it is moot whether the presumed antenatal magnesium deficiency might have been corrected.
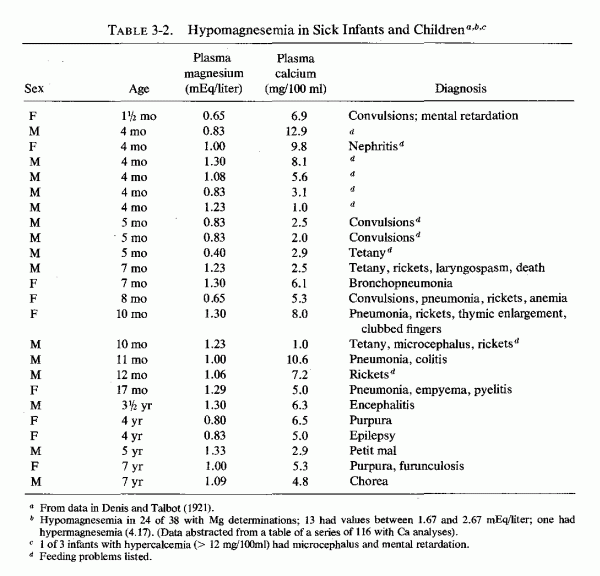
Magnesium determinations during infancy have not been frequently reported; when reported, they have rarely included data on the maternal or infant status (Table 3-1) and (Table 3-1 continued). The first report found was one in which 24 magnesium levels were included in a table of 116 infants and children with a variety of abnormalities whose calcium levels had been analyzed (Denis and Talbot, 1921; Table 3-2). When the syndrome of magnesium malabsorption was recognized and infantile hypocalcemia was found often to be unresponsive to calcium or calcemic agents but to respond to magnesium repletion, magnesium determinations were done more commonly. The change in infant feeding patterns from breast-feeding to use of a variety of formulas has led to increased mineral retentions that are not paralleled by calcium and magnesium plasma levels, which are lower in infants fed cows' milk than in normal infants who are breast-fed.
Infants at greatest risk of neonatal hypomagnesemia are low-birth-weight infants, including those suffering from intrauterine growth retardation (IUGR) or premature infants recovering from birth hypoxia or later respiratory distress, and infants born to very young primiparous women or to young mothers who have had frequent pregnancies or multiple births, to preeclamptic mothers, and to diabetic mothers. Plasma magnesium levels are a less reliable index of magnesium deficiency than is the parenteral load test, and magnesium deficiency, so demonstrated, has been found to be more common in newly born premature than in full-term infants, even when not indicated by notable hypomagnesemia (Caddell, 1975). The incidence of neonatal magnesium insufficiency may be greater than suspected. The tendency of women with preeclampsia or eclampsia to develop rising plasma magnesium levels during the last month of pregnancy, even without magnesium therapy, despite which they retain high percentages of parenterally administered pharmacologic doses of magnesium, suggests that magnesium deficiency might be far more common during pregnancy than is indicated by the incidence of hypomagnesemia.
3.1. Magnesium Deficiency during Gestation
3.1.1. Effects of Experimental Maternal Magnesium Deficiency on the Fetus
To attribute the high incidences of placental and fetal abnormalities, stillbirths, and neonatal deaths (found among infants born to eclamptic women) to magnesium deficiency during gestation would be highly speculative at this stage of our knowledge. However, there are provocative findings that point to the possibility that it is likely to be contributory, not only to complications of pregnancy, but to damage to the products of conception. Interrelationships with other factors must be considered.
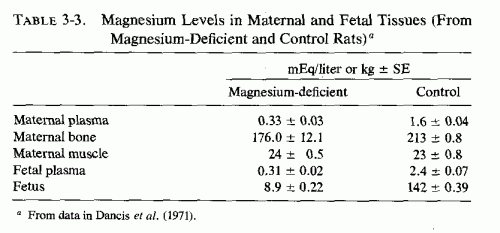
Rats kept severely magnesium depleted (receiving 1/200 the control magnesium intake) for the entire 21-day period of gestation had no living fetuses at term (Hurley and Cosens, 1970, 1971; Hurley, 1971; Hurley et al., 1976). The shorter the duration of the magnesium deficiency, the fewer implantation sites were affected. When the deficiency was maintained from day 6-12, about 30% of the implantation sites were involved and 14% of the full-term fetuses had gross congenital abnormalities (cleft lip, hydrocephalus, micrognathia or agnathia, clubbed feet, adactyly, syndactyly, or polydactyly, diaphragmatic hernia, and heart, lung, and urogenital anomalies). Milder magnesium deficiency (1/130 control intake) maintained throughout pregnancy resulted in resorption of half the implantation sites and malformation of the living young at term. In addition to their congenital anomalies, the surviving young were anemic and edematous. Surprisingly, despite the marked fetal damage caused by the deficiency during gestation, the pregnant rats showed only mild signs of magnesium deficiency despite sharp drops in their plasma magnesium levels. The severity of fetal damage produced in these studies was greater than in other studies; there might have been concomitant trace element deficiencies. Magnesium deficiency (1/130 of control intake), comparable to that produced in the less severely depleted rats of Hurley et al. (1976), but produced by adding salt mixtures with only the magnesium contents differing, resulted in less severe damage (Dancis et al., 1971; Cohlan et al., 1970). When the magnesium-deficient diet was fed from the second day of gestation to term, only one of eight rats bore a litter; the remainder had evidence of resorption at implantation sites. When the magnesium-deficient diet was fed from ninth or tenth day to term, the magnesium-deficient rats all produced live litters (8.1/litter), but there were also 36 resorption sites among the 17 test rats. The control rats had no resorption sites and delivered 8.5 pups/litter. The pups born to deficient dams were small (2.6 ± 0.1 g, in comparison to control mean weight of 3.8 ± 0.3 g) and were weak and pale. There was consistent microcytic anemia; edema was prominent in the severely anemic fetuses. The control fetuses had higher plasma magnesium levels than did the fetuses of the magnesium-deficient rats, a finding suggesting that there is relatively little protection of the fetus against maternal magnesium deprivation. The mothers looked healthy at term, and although they had hypomagnesemia, their tissue magnesium levels were only slightly lowered. In contrast, the fetal tissues were markedly magnesium depleted (Table 3-3). There was little difference in placental magnesium in the control and deficient groups, but the placental calcium of the magnesium-deficient fetuses also had higher tissue calcium levels (105 as compared with 80.1 in control fetuses).
The less severe magnesium-depletion gestation study of Wang et al. (1971), which provided 1/10 the control amount of magnesium to deficient rats, did not significantly reduce the number of offspring but markedly reduced their viability. Labor was prolonged in the depleted group, and 53 (36%) of the 146 offspring were stillborn. By the fifth day after birth, 82 more had died spontaneously or been eaten by their mothers; only 7.5% survived. There were no obvious abnormalities, other than small size and occasional swelling of extremities. The deficient mothers were normal in weight but had significantly lower-than-control levels of serum and bone magnesium. They also exhibited impaired lactation, and secreted milk significantly lower in magnesium than that of control rats. The survival of pups fed by the magnesium-deficient dams was poor.
Dancis et al. (1971) speculated that the higher placental and fetal calcium levels of the magnesium-deficient rats might have reflected increased fetal parathyroid activity.
3.2. Perinatal Parathyroid Secretion: Interrelations with Magnesium and Calcium
3.2.1. Hyperparathyroidism of Pregnancy
There is mounting evidence of magnesium insufficiency during pregnancy. Experimental acute magnesium deficiency has caused increased parathyroid secretion and even parathyroid hyperplasia (Larvor et al., 1964a; Kukolj et al., 1965; Gitelman et al., 1965, 1968a,b; Lifshitz et al., 1967; Sherwood et al., 1970, 1972; Targovnik et al., 1971). Thus, the possibility that magnesium deficiency is contributory to hyperparathyroidism of pregnancy, which is common despite widespread supplementation with calcium and vitamin D at that time, should be considered.
Low normal or subnormal plasma phosphorus levels during pregnancy, which rise postpartum, have long been associated with maternal hyperparathyroidism (Mull and Bill, 1934, 1936; Mull 1936; Bodansky, 1939). This condition has been found so frequently as to be termed "physiologic" (Hamilton et al., 1936; Cushard et al., 1972). Rat studies have shown that pregnancy can cause significantly increased parathyroid volume (Opper and Thule, 1943). Significant maternal hyperparathyroidism has been demonstrated by immunoreactive parathyroid hormone (PTH) measurements, the levels of PTH being significantly higher during the third trimester and at delivery than in age-matched nonpregnant women and than in cord blood (Samaan et al., 1973). Samaan et al. (1973) suggest that maternal hyperparathyroidism might be a response to the high fetal needs for calcium during the third trimester.
Despite hyperparathyroidism, serum calcium and magnesium levels both tend to be subnormal, especially during the third trimester of pregnancy (Watchorn and McCance, 1932; Mull and Bill, 1934; Mull, 1936; Bodansky, 1936; Kerr et al., 1962; Newman, 1957; DeJorge, 1956b; Lim et al., 1969b), which suggests that the gestational hyperparathyroidism can be secondary to hypocalcemia and/or to hypomagnesemia rather than physiological. Hyperparathyroidism has been found in mothers of infants with neonatal hypomagnesemia and hypocalcemia (J. A. Davis et al., 1965; Ertel et al., 1969; Monteleone et al., 1975).
Ludwig (1962) reviewed the relationship of hyperparathyroidism to gestation and the products of conception, and found that there was a greatly increased incidence of complications of pregnancy and of fetal loss and infant morbidity among diagnosed hyperparathyroid women. Since asymptomatic hyperparathyroidism (with gestation-hypocalcemia) is common during even normal pregnancy and has been implicated in symptomatic infantile hypocalcemia, the possibility should be considered that there might be a common denominator that contributes to both. The importance of calcitonin secretion, both in the mother and the neonate, is gaining increasing recognition. The role of magnesium deficiency during gestation should also be considered, since it is involved in parathyroid dysfunction and in calcitonin secretion. Maternal abnormalities that predispose to neonatal convulsive hypomagnesemic hypocalcemia include maternal magnesium deficiency, which predisposes to maternal hyperparathyroidism and is apt to occur in: (1) adolescent or young mothers (whose own magnesium requirements may not be fully met); (2) preeclamptic or eclamptic women; (3) women who have had several pregnancies in rapid succession, or with multiple births; (4) mothers with malabsorption; and (5) women with diabetes mellitus (Reviews: Tsang and Oh, 1970a; Tsang, 1972; Tsang and Steichen, 1975; Tsang et al., 1977a,b). Intrinsic (pregestational) hyperparathyroidism, of course, falls into this category.
The possibility that magnesium deficiency of pregnancy might be contributory to both transitory and sustained maternal hyperparathyroidism (with low serum calcium levels) should be considered, and the response to magnesium administration investigated.
3.2.2. Fetal Parathyroid Activity and Phosphate, Calcium, and Magnesium Homeostasis
The fact that cord blood phosphate, magnesium, and calcium levels are usually higher than maternal levels (Bakwin and Bakwin, 1932; Finola et al., 1937; Bruck and Weintraub, 1955; Delivoria-Papadopoulos, 1967; Samaan et al., 1973; Bergman, 1974; David and Anast, 1974; Tsang et al., 1973b, 1976b) suggests that fetal homeostasis of these elements is at least partially independent of maternal factors. Maternal hyperparathyroidism has long been speculated to be a direct or indirect cause of neonatal hypoparathyroidism, which contributes to hyperphosphatemia and secondary hypocalcemia and hypomagnesemia that are seen in the early days to weeks of life (Friderichsen, 1938, 1939; Van Arsdel, 1955; Hutchin and Kessner, 1964; Hartenstein and Gardner, 1966; Mizrahi et al., 1968; Ertel et al., 1969; Tsang et al., 1973a). Severe enough experimental hyperparathyroidism in pregnant rats, however, to cause hypercalcemia and renal and myocardial damage in the mothers caused no more fetal hypercalcemia than was seen in control fetuses and caused no fetal soft tissue calcinosis (Krukowski and Lehr, 1961a,b; Lehr and Krukowski, 1961), suggesting that the placental barrier protected the fetus against maternal hyperparathyroidism. These investigators reviewed the literature at that time, and discussed the early experimental evidence that PTH does not penetrate the placental barrier, either from the maternal to the fetal circulation, or from the fetus to the mother. Earlier, Hoskins and Snyder (1933) showed that injection of PTH into the dog fetus in utero resulted in elevated fetal serum calcium levels not associated with a simultaneous rise in maternal plasma calcium levels. PTH injection into the pregnant dog raised maternal but not fetal calcium levels. An accidental finding during another study of the effect of hyperparathyroidism in dogs was obtained when one of the dogs was found to be pregnant (Cantarow et al., 1938). The absence of damage to the fetuses, such as had been produced by PTH in the mother, was interpreted as indicating possible lack of passage of PTH through the placental barrier. When they confirmed these findings in their own controlled experiments with rats (Lehr and Krukowski, 1961; Krukowski and Lehr, 1963), they judged that since the placental membrane is at least three cell layers thick (Wislocki and Dempsey, 1955) even at the time of maximal placental permeability, large proteins such as PTH are unlikely to penetrate it. This hypothesis has been proved correct. Garel and Dumont (1972) have shown no demonstrable maternal-fetal or fetal-maternal crossover of tagged PTH in the rat. Injection of PTH to fetal rats has raised their serum calcium levels, and influenced their serum magnesium and phosphate levels (Garel, 1971b; Garel and Barlet, 1974). The effect of PTH injections into the rat fetus suggests that it mobilizes bone calcium, as indicated by exposure of fetal rat bones to PTH (Raisz and Niemann, 1967, 1969). Garel and Barlet (1974) were unable to confirm earlier observations that PTH decreases fetal plasma phosphate (Garel and Geloso-Mayer, 1971), and speculated that mobilization of bone mineral by FTH should increase fetal plasma phosphate levels.
In contrast to the inability of maternal PTH to cross the placenta, calcium and magnesium are readily transferred across the placental barrier (MacDonald et al., 1965). Their higher levels in fetal than in maternal blood suggest that there is active placental transport from maternal to fetal circulation (Economu-Mavrou and McCance, 1958; Aikawa and Burns, 1960; Cohlan et al., 1970). An active placental transport mechanism involving calcium and magnesium-stimulated ATPase has been identified (Whitsett et al., 1977a,b).
Inferential evidence has been obtained that modulation of increased or decreased fetal parathyroid activity protects the fetus against maternal hyper- or hypocalcemia and hyper- and hypophosphatemia, whether induced by dietary means, by maternal parathyroidectomy (Sinclair, 1942), high doses of PTH (Lehr and Krukowski, 1961; Krukowski and Lehr, 1963), or by hypervitaminosis D (Potvilege, 1962). The same should be true for protection against hyper- or hypomagnesemia. More recently there has been experimental proof that fetal parathyroids are functional. Garel and Geloso-Meyer (1971) demonstrated that thyro- or parathyroidectomy of pregnant rats causes fetal as well as maternal hypocalcemia, secondary fetal parathyroid hyperplasia, and resultant rises in the fetal plasma calcium levels. Ablation of the fetal parathyroids or injection of anti-PTH serum into the rat fetus (Garel, 1971a) has resulted in sustained fetal hypocalcemia. Production of fetal hypocalcemia by injection of the disodium salt of EDTA (which also chelates magnesium, although this was not measured) into sheep fetuses caused increased fetal PTH levels, but no change in the maternal PTH levels. In contrast, infusion of EDTA to normocalcemic ewes in late pregnancy caused a marked reduction in maternal plasma unchelated calcium and a doubling of maternal PTH levels, but no significant change in either of these parameters in the fetuses. Infusion of calcium to the pregnant ewes lowered their PTH levels but caused no change either in calcium or PTH levels of their fetuses (Care et al., 1975). Studies in monkeys, however, have shown that fetal serum PTH was undetectable in the basal state and in response to EDTA-induced fetal hypocalcemia, although EDTA-induced maternal hypocalcemia caused 30-197% increases in maternal PTH values (A. R. Fleischman et al., 1975). Whether the presumed simultaneously reduced serum magnesium levels interfered with release of PTH from the fetal glands requires investigation. Garel and Barlet (1976) have shown species differences in the parathyroid status at birth. Thus, there should be caution in applying experimental findings to human perinatal hormone/mineral interrelationships.
Much less work has been done on the fetal parathyroid response to low magnesium levels. Since fetuses of magnesium-deficient rodents show more damage than do the mothers, it seems likely that fetal parathyroid activity is less effective in protecting the fetus against hypomagnesemia than against hypocalcemia.
3.2.3. Hypoparathyroidism of Infancy
Hyperparathyroidism of pregnancy has long been blamed for hypoparathyroidism and low serum calcium/phosphorus ratios in the neonatal period (Friderichsen, 1939; Bakwin, 1939). The existence of neonatal tetany is considered a sensitive clue to maternal hyperparathyroidism. Hartenstein and Gardner (1966) reviewed the literature and found that there were seven reported families, including their own reports, in which neonatal tetany was associated with maternal parathyroid adenoma. Friderichsen (1939) was the first to report the association in an infant who developed infantile tetany at five months of age, and whose mother had osteitis fibrosa cystica secondary to her parathyroid adenoma. Brief reference was made to unusually severe signs of hypocalcemic neonatal tetany on the second day of life of two infants born to hyperparathyroid mothers (Talbot et al., 1954). Maternal symptoms can well be absent in hyperparathyroid mothers whose premature or full-term infants present with severe tetany (Walton, 1954; Van Arsdel, 1955). A mother of eight children (four of whom had had hypocalcemic neonatal tetany developing at the 14th, 12th, 9th, and 2nd days) who had another pregnancy that aborted had asymptomatic parathyroid adenoma that was not diagnosed until her renal calcinosis was found a year and a half after the birth of her last child (Hutchin and Kessner, 1964). Conversely, infantile hypocalcemic tetany did not develop until one year of age in an infant, three months after cows' milk was substituted for breast milk, which had been provided by his mother who had had symptoms and signs of hyperparathyroidism (Bruce and Strong, 1955). Hypoparathyroidism was diagnosed in that child in his fourth year of life; a parathyroid adenoma was removed from the mother six years after he was born.
It was first suggested by Pincus and Gittleman (1925) that transient hypoparathyroidism might be at fault in a seven-week-old infant with nonrachitic tetany. Bakwin (1937) considered the susceptibility of neonatal infants to hyperphosphatemia and secondary hypocalcemic tetany to be a result of the phosphate load provided by cows' milk fed to infants with end-organ unresponsiveness to PTH at birth. However, fetal plasma phosphorus levels are usually considerably higher than are maternal plasma levels (McCance and Widdowson, 1954, 1961), and even breast-fed infants who do not have a free supply of milk during the first 48 hours show a rise in serum inorganic phosphate after the first 24 hours (McCance and Widdowson, 1961). This rise has been attributed to the expenditure of tissue glycogen and protein to maintain life while the intake is minimal, an observation that has been supported by study of fasting newborn pigs (McCance and Widdowson, 1957). Before and at birth (cord blood) there are elevated fetal or infant plasma phosphorus levels that are associated with higher than maternal plasma levels of calcium and magnesium (Reviews: Smith, 1959; Bergman, 1974; Tsang et al., l976b).
3.2.3.1. Hypocalcemia of Infancy
A few hours after birth, infants commonly exhibit sharp drops in plasma calcium levels (Review: L. Bergman, 1974). Their phosphate levels tend to remain high for days to weeks, especially those fed cows' milk. This is seen in normal full-term infants but is particularly marked in low-birth-weight infants, those that are born to diabetic mothers, or those that have been born after difficult deliveries and suffered birth hypoxia or later respiratory distress. It has been stressed that early neonatal hypocalcemia should be distinguished from that developing only after a week of life or later, which is related to the phosphate load of cows' milk. The foregoing section on neonatal and persistent infantile hypoparathyroidism [particularly with reference to the four siblings who developed the syndrome at 2-14 days (Hutchin and Kessner, 1964)] suggests that there might be a common denominator for both, and that the phosphate load precipitates the syndrome in less abnormal infants.
The greater predilection for hypocalcemia and hyperphosphatemia among premature than full-term infants, and the rarity with which breast-fed infants develop these abnormalities within the first three weeks of life, were clearly depicted by Bruck and Weintraub (1955). Both groups had lower calcium levels after birth than they had had in their cord blood. Few of the premature hypocalcemic infants had tetanic symptoms; they more commonly presented with convulsions, hypersensitivity, rigidity, edema, vomiting, respiratory disturbances, and drowsiness. However, there were frequently no abnormal symptoms. The authors cautioned against considering asymptomatic hypocalcemia as "physiologic," since sudden transition from latent to manifest tetany is frequent. In the 1918 review of infantile tetany by Howland and Marriott, they reported four publications on the syndrome from 1815 through 1887. They were the first to observe that the syndrome could occur in the absence of rickets, and that it was far more common in cows'-milk-fed than in breast-fed infants. Dodd and Rapaport (1949), in their review, reported only sporadic cases from 1913. Among their own series of 33 infants with symptomatic neonatal hypocalcemia, 22 had convulsions, 28 had vomiting, 16 had edema (severe in 9), and 12 were cyanotic. Hemorrhagic manifestations included hematemesis (6 cases), melena (4), and hemoptysis or petechiae (2). Saville and Kretchmer (1960) commented on the rarity of reports of neonatal tetany until late in the 1930s, and its increasing frequency thereafter. They reviewed the evidence that a combination of cows' milk and vitamin D supplementation, together, were potent means of inducing infantile hypocalcemia and considered the high incidence in the literature among infants born after difficult labor or to diabetic mothers. They confirmed these observations in their series of 125 cases in a major medical center from 1940 to 1958. Only 33% were the products of normal full-term pregnancies and uneventful labor. Almost a tenth were born to diabetic mothers. Both low-birth-weight infants and those born to diabetic mothers, as well as other "sick" and hypocalcemic infants, have been shown to have subnormal parathyroid function (L. David and Anast, 1974; Samaan et al., 1973; Tsang et al., 1973b, 1975a, 1976a, 1977a; Bergman, 1974; Bergman et al., 1974; David et al., 1976, 1977).
It has been speculated that the hypoparathyroidism of infancy might be related to parathyroid immaturity (especially in premature or dysmature infants), to functional parathyroid deficiency, or to fetal hypercalcemia, possibly deriving from maternal hyperparathyroidism-induced hypercalcemia that might cause fetal PTH suppression, mediated by resultant fetal hypercalcemia (Reviews: Tsang et al., 1973a, 1976a).
On the basis of the early experimental evidence as to fetal parathyroid competence, Lehr and Krukowski (1961b, 1963) commented that it is invalid to blame the neonatal rise in serum phosphate, with resultant drop in serum calcium, on the inability of functionally immature neonatal parathyroids to compensate for the loss of maternal PTH. They suggested that the difference between maternal and fetal serum calcium levels might reflect the higher pCO2 in fetal blood, a deduction made on the basis of their observation that hypoxic fetuses (taken from dams after sacrifice) had markedly higher serum calcium levels than did fetuses without hypoxia (taken from living anesthetized dams) (Krukowski and Lehr, 1963). They proposed that the drop in serum calcium to normocalcemic levels immediately after birth might be mediated by initiation of respiration with blowing off of excessive CO2 It is of interest, in this regard, that correction of neonatal acidosis by administration of bicarbonate in premature infants (Tsang and Oh, 1970a; Tsang et al., 1976b), in infants with intrauterine growth retardation who often have asphyxia (Tsang et al., 1975a), and in infants (often with respiratory distress) of diabetic mothers (Tsang et al., 1974) results in further reduction in serum calcium, with production of continued hypoparathyroidism. These findings call to mind the hypothesis of Barzel (1971) that PTH function is influenced by the bicarbonate/carbonic acid buffer system. He has shown that hypoparathyroid patients have simultaneously elevated plasma phosphate and pCO2 levels, with normal blood pH. However, the persistence of hypoparathyroidism, despite both hyperphosphatemia and hypocalcemia in infants whose elevated pCO2 and acidosis have subsided, suggests that another mechanism can be operative. It is unlikely to be neonatal parathyroid immaturity; fetal parathyroid function has been shown to protect the fetus against experimental maternal aberrations in phosphate, calcium, and magnesium levels; and immunoreactive evidence of fetal PTH has also been obtained (supra vide).
Nonetheless, low plasma PTH levels have been demonstrated in the first day or two of life (Tsang et al., 1973b; Samaan et al., 1973; L. David and Anast, 1974; Root et al., 1974; Tsang et al., 1975a), indicating failure of PTH secretion or release during the early neonatal period. Tsang et al. (1973b) have shown that PTH levels did not increase during the 24- to 48-hour period during which serum calcium levels fell. They found no relationship between serum PTH levels and total and ionized calcium in maternal, cord, and infant sera. Less gestationally mature infants had less increase in serum PTH during hypocalcemia than did the more mature infants. In contrast, David et al. (1976, 1977) showed that low-birth-weight infants had higher immunoreactive (i) PTH levels at birth than did normal adults, and that the iPTH levels increased earlier and were higher than in normal full-term infants (David and Anast, 1974; David et al., 1977). These investigators commented on the difference between their findings and those of Tsang et al. (1973), whose study infants were more severely hypocalcemic than were those of David et al. (1977). Additionally, the infants in the French study (David et al., 1977) were breast-fed; those in the American study (Tsang et al., 1973b) were bottle-fed. Possibly the absence of hyperphosphatemia and hypomagnesemia in the French infants might reflect the difference in the feeding customs. It is conceivable that the rise in PTH in premature rhesus monkeys as early as six hours after delivery (Fleischman et al., 1975) might be similarly explained.
3.2.3.2. Magnesium Deficiency and Infantile Hypoparathyroidism
The evidence that magnesium deficiency during gestation and in the neonatal period can be correlated with parathyroid dysfunction suggests that magnesium deficiency might well be an important contributory factor to infantile hypoparathyroidism, failure of target organ response to PITH, and to hypocalcemia. Inadequate supply of magnesium to the fetus can result from insufficient maternal intake, abnormalities of pregnancy during which there is subnormal maternal magnesium or placental damage that interferes with transport of nutrients including magnesium to the fetus. High-risk infants, usually born to mothers with abnormalities of pregnancy, have a high incidence of transient or prolonged hypoparathyroidism with symptomatic neonatal hypocalcemia. Their magnesium deficiency is usually detected later, either as hypomagnesemia, often after calcemic agents have failed to control neuromuscular irritability, or by demonstration of high percentage retention of parenteral loads of magnesium.
Hypoparathyroidism was reported in two infant sisters (children of first cousins), in association with severe hypomagnesemia (0.5 and 0.4 mEq/liter, respectively) that was detected subsequent to treatment of their hypocalcemia with high doses of vitamin D (100,000 U/day) or dihydrotachysterol (Niklasson, 1970). Despite the calcemic agents, their serum calcium levels rarely reached normal or hypercalcemic levels. One exhibited mental retardation and emotional lability at 20 months of age. Convulsions were common in this family, a finding that suggests that there may have been a genetic abnormality in magnesium metabolism. The correlation of maternal magnesium deficit with maternal hyperparathyroidism, and with neonatal hypoparathyroidism and hypomagnesemic hypocalcemic tetany and convulsions, is inferential evidence that the infants reported by Niklasson (1970) are not likely to be unique. David and Anast (1974) showed immunoreactive PTH levels to be low during the first nine days of life in normal, "sick," and hypocalcemic infants. They found that depressed plasma magnesium levels (range = 0.97- 1.25) were frequent (20%) in hypocalcemic infants. In normal newborn infants the range of plasma magnesium was 1.6-1.75 mEq/liter. These infants' hypomagnesemia was transient, usually reaching normal levels even when magnesium supplements were not given, or when the hypocalcemia was corrected by treatment with calcium. The rarer but more severe form of neonatal hypomagnesemic hypocalcemia associated with magnesium malabsorption must be treated with large magnesium supplements for correction of parathyroid suppression.
However, an infant has been described with the same syndrome but with hyperparathyroidism (Monteleone et al., 1975). The authors suggested that his seizures, which were intensified by calcium but responded to magnesium therapy, might have been causally related to hypomagnesemia secondary to his mother's hyperparathyroidism. They regretted that the PTH determination had been run after magnesium treatment had been started, thereby making it impossible to rule out the possibility of functional hypoparathyroidism immediately after birth. The infant's continued hypocalcemia and elevated PTH values suggested that he might have had target organ unresponsiveness to PTH, such as has been reported in magnesium-depleted older patients. They referred to the suggestion of L. Chase et al. (1974) that hypomagnesemic patients with hypocalcemia might have impaired skeletal response to PTH, with decreased heteroionic exchange of magnesium at the bone surface, a hypothesis proposed also by Zimmet (1968), who cited Neuman and Neuman (1957) regarding the theory that cation exchange for calcium occurs predominantly at the hydration shell.
3.3. Calcitonin during Gestation; Interrelations with Magnesium and Calcium
3.3.1. Calcitonin during Pregnancy
Calcitonin (CT) levels are higher in maternal blood at time of delivery than they are in nonpregnant women (Samaan et al., 1973a,b, 1975). Pregnant ewes have elevated CT levels during the last 40 days of gestation, both on a low- and high-calcium intake (Barlet, 1974; Barlet and Garel, 1974; Garel et al., 1974, 1976; Garel and Barlet, 1975). Since the highest levels have been found in the ewes bearing triplets, there is support for the suggestion (Lewis et al., 1971) that CT might function to protect the bones of pregnant or lactating females against excessive demineralization (by the increased PTH of pregnancy) to meet fetal calcium needs. The response to hypercalcemia in pregnant animals is increased CT secretion. Infusion of calcium salts has augmented the CT secretion of pregnant ewes (Garel et al., 1973, 1974, 1976). Since pregnant women characteristically have hyperparathyroidism with hypocalcemia, as well as hypomagnesemia, the CT-stimulatory mechanism would appear not to be hypercalcemia. The hypocalcemia might reflect the response to CT secretion that spares the maternal skeleton. The mechanism by which CT secretion is increased in the presence of hypocalcemia, which (from the above studies) should decrease C-cell activity, remains unclear. Possibly, the simultaneously low magnesium levels during pregnancy play a role. For example, although magnesium-deficient rats show increased C-cell activity and release of CT in the presence of hypercalcemia (Stachura and Pearse, 1970), magnesium-deficient dogs with hypocalcemia also develop C-cell hyperplasia and evidence of increased secretory activity (Rojo-Ortega et al., 1971, 1971/1973). Thus, during human pregnancy, when plasma levels of both calcium and magnesium are low, conflicting responses might be responsible for both hyperparathyroidism and hypercalcitoninemia.
3.3.2. Fetal Secretion of Calcitonin
Maternal CT has been shown not to cross the placental barrier in rats (Garel et al., 1969, 1973, 1976) or in ewes (Garel et al., 1974). Fetal thyroid tissue is able to secrete CT, which exerts a hypocalcemic effect (Garel et al., 1968, Garel, 1969). That fetal C-cells can respond to hypercalcemia has been shown by Littledike et al. (1972) and Garel et al. (1973, 1974), who evoked significant increases in plasma CT in ovine, bovine, and porcine fetuses by acute elevations in fetal calcium levels, Administration of exogenous CT to the rat fetus, late in gestation, lowers the plasma levels of all three elements; calcium, magnesium, and phosphorus (Garel et al., 1968, 1969; Garel and Barlet, 1974). Samaan et al. (1975) attribute infantile hypocalcemia to elevated CT levels.
3.3.3. Neonatal Calcitonin
The level of immunoreactive CT (iCT) is significantly higher in the cord blood of full-term infants than in maternal blood at time of delivery following normal pregnancies (Samaan et al., 1973a,b, 1975). By use of a method of determination that does not detect iCT in normal children and adults (150 pg/ml), David et al. (1977) found just detectable levels in cord blood of low-birth-weight infants, with a marked increase after 1-2 hours of age, and a peak almost 5-fold higher by 11 hours after birth. Similar findings were reported in infants of diabetic mothers (Bergman, 1974; Bergman et al., 1974). Severalfold-higher plasma CT levels have also been detected in newborn lambs than in their mothers (Garel et al., 1974), and the levels have risen in response to calcium per os (Garel et al. 1976) and in response to injection of cholecystokinin-pancreozymin (Barlet and Garel, 1976). Garel (1969) demonstrated that injection of CT into newborn rats produced a marked lowering of their plasma calcium levels. On the other hand, in subsequent work showing species differences in PTH/CT/Ca/Mg interrelationships in newborn ruminants, Garel and Barlet (1976) pointed out that the CT levels do not necessarily correlate with plasma calcium levels. Bergman (1974) postulates that high levels of growth hormone (i.e., in response to glucose infusion) at the time that CT levels are high increases the risk of neonatal hypocalcemia. Samaan et al. (1975) attribute infantile hypocalcemia to elevated CT levels.
The high blood levels of CT of neonates, and the preliminary evidence that CT secretion is increased in magnesium depletion (Stachura and Pearse, 1970; Rojo Ortega et al., 1971), as well as in the presence of high magnesium levels (Radde et al., 1970; Care et al., 1971; Littledike and Arnaud, 1971; S. P. Nielsen, 1971/1973, 1974; Barlet et al., 1974), suggests that early and sustained infantile hypocalcemia might be a function of combined hypomagnesemia/hypoparathyroidism/increased CT secretion-all of which respond to moderate doses of magnesium. The possibility that perinatal magnesium deficiency might be a contributory or even a fundamental abnormality in the mineral and hormonal aberrations of the perinatal period has received little consideration.
3.4. Perinatal Hypervitaminosis D
3.4.1. Toxicity of Excess Vitamin D during Pregnancy
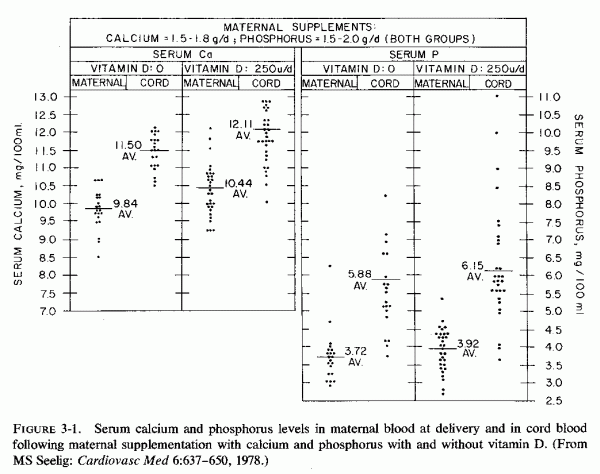
In the late 1930s, the decade that vitamin D supplementation became fairly commonplace, placental scarring and calcification were found to be more common in women supplemented with viosterol (vitamin D than in those who were not supplemented or who were given cod liver oil (vitamin D + vitamin A) (Brehm, 1937). The same year, Finola et al. showed that viosterol (vitamin D 250 U/day) given with calcium phosphate supplements caused little or no change in serum phosphorus levels as compared with the levels of those given the calcium salt alone, but several had serum calcium levels at or above 11 mg/100 ml (Fig. 3-1). Cord blood analyses showed a shift toward higher phosphorus levels and a higher incidence of hypercalcemia among infants of mothers given viosterol plus calcium phosphate than among those born to mothers on the calcium diphosphonate alone, although the averages were similar (Fig. 3-1). Both groups (Finola et al. 1937; Brehm, 1937) expressed concern about the tendency toward intrauterine osteosclerosis in the infants of the vitamin-D-supplemented mothers, which was associated with narrowed and closed fontanels, a finding they considered contributory to longer, more difficult labors. Of greater concern to Brehm (1937) was the placental calcification, scarcely notable in those who had not had vitamin D supplementation, but so marked among several of the women given viosterol plus calcium as to interfere with placental separation. Three stillborn infants with severe renal calcification were born to that group of mothers. No note was taken in either of these studies of the maternal intake or levels of magnesium, but studies of customary magnesium intakes at about that time suggest that intakes might not have been optimal. These are preliminary observations that should be tested, a difficult undertaking with American women because milk in the United States is almost universally fortified with vitamin D2 (400 units/quart) and calcium and vitamin supplements, providing 400 IU of vitamin D per tablet, are given to most pregnant women. This practice has been widespread since it was realized in the 1930s that failure to meet the demands for calcium, which increase manyfold during the last trimester, can cause maternal hypocalcemia (Cantarow et al., 1938). The need for prenatal vitamin D supplementation was predicated on the observation of rickets of the newborn (Coons and Blunt, 1930).
Subsequent work suggests that this practice might not be uniformly beneficial. Low magnesium intakes, such as are common during pregnancy, in combination with calcemic agents, favor a high Ca/Mg ratio. Experimental studies show that high Ca/Mg and Na/K ratios increase arterial resistance (Review: Haddy and Seelig, 1976/1980. Whether the high Ca/Mg ratio of intake during pregnancy might contribute to toxemic hypertension must be explored. Furthermore, normal and abnormal vitamin D metabolites have widely differing potency and toxicity (Seelig and Mazlen, 1977). Whether subjection of foodstuffs, to which vitamin D has been added, to a variety of cooking processes might convert the antirachitic factor to more toxic derivatives has not been investigated. It is known, for example, that peroxidized cod liver oil and some of its fractions can damage the placenta, with resultant intravascular coagulation and eclampsia in rats (McKay, 1962; Kaunitz et al., 1962, 1963; McKay and Goldenberg, 1963; McKay and Wong, 1962; McKay et al., 1967). Also, administration of high doses of even untreated vitamin D to rats has caused decreased placental volume, with atrophy and mucoprotein infiltration in the portion of the placenta composed of allantoic villi carrying fetal vessels (Potvliege, 1962). The fetal capillaries show degeneration of the endothelial cells, and the intervillous spaces collapse and become relatively bloodless. Calcium deposition occurs late in degenerated villi and in the walls and surrounding mesenchyma of large fetal vessels. Ornoy et al. (1968) has also shown that hypervitaminosis D in rats causes decreased placental volume. The young of the hypervitaminotic rats with placental pathology are small for gestational age, a finding similar to that seen in human infants born to eclamptic mothers or to others with abnormal placentas. Potvliege (1962) has found that there is also significant decrease in the volume of the parathyroids both in dams and fetuses, suggesting that the vitamin D might have caused hypercalcemia in both. The mothers had marked systemic calcinosis. The fetuses, however, showed neither vascular lesions nor excessive calcium deposition. In fact, pregnant rats that developed placental lesions (Ornoy et al., 1968) had fetuses with defective bone formation. These investigators attribute the anomalous bone formation to damage to fetal osteogenic tissues induced by passage of excessive vitamin D2 through the vitamin-impaired placenta. They speculate that vitamin D-impaired placental function permits excessive vitamin D to reach the fetuses, and presume that fetal damage is caused by vitamin D2 toxicity at the cellular level. W. Friedman and Roberts (1966) have shown that the blood levels of antirachitic sub stance are high both in rabbit mothers given toxic amounts of vitamin D2 and their young, but the fetal damage produced resembles more that seen in human babies during the epidemic of infantile hypercalcemia during a time of excessive vitamin D prophylaxis of rickets (Reviews: Black, 1964; Seelig, 1969b) than that seen in the rats. As with the rats, the does poisoned with vitamin D2 had greater damage than did their young, but the offspring had cardiovascular lesions: supravalvular aortic stenosis (Coleman, 1965; Friedman and Roberts, 1966; Friedman, 1968), endocardial thickening (Coleman, 1965), and premature closure of the fontanels, osteosclerosis, and palatal abnormalities (Friedman and Mills, 1969). It seems likely that both vitamin D3 and its 25-hydroxy-derivative cross the placental barrier from mother to fetus in the rat (Haddad et al., 1971). Judging from comparable 25-OH-D3 levels in maternal and cord blood, placental transport probably also takes place in humans (Hillman and Haddad, 1974; Belton et al., 1977). The observation that levels of 25-OH-D3 are lower in newborn rabbits with supravalvular aortic changes, born of does with hypervitaminosis D than they are in controls (Mehlhorn et al., 1977) suggests that administration of toxic amounts of vitamin D might result in its abnormal metabolism. It can be speculated that, as the enzymes involved in normal vitamin D metabolism are overloaded, abnormal degradation products can be produced. Whether there are such abnormal products, and whether they are more toxic than the normal metabolites should be investigated.
Unfortunately, although the enzyme systems involved in hepatic and renal hydroxylation of vitamin D are magnesium dependent, magnesium levels have not been determined in the studies of vitamin D toxicity in pregnancy, nor in the dam aged young. Since administration of high doses of magnesium is protective against vitamin D toxicity and magnesium deficiency intensifies the damage produced, the interrelationships of vitamin D and magnesium during pregnancy should be studied. Does magnesium deficiency increase the risk of vitamin D toxicity, and if so, to what extent? This is a cogent point, since the average American woman probably ingests considerably more than optimum quantities of vitamin D from fortified milk and other foods, as well as from prenatal supplements. Her intake of magnesium is likely to be marginal, at best, and is likely to be significantly low. Can magnesium supplements during pregnancy protect against vitamin D toxicity, and to what extent? This question might be relevant to protection against eclampsia, damaged placenta, and intrauterine growth retardation, as well as against fetal abnormalities-from bone to renal to cardiovascular anomalies-such as have been seen in experimental vitamin D toxicity during pregnancy, and some of which have been related to experimental magnesium deficiency itself. The nature of the fetal abnormalities caused by experimental hypervitaminosis D during gestation seems not be a function of the vitamin D alone, but to other components of the diet in ways that have not yet been clearly defined. The early study by Nicholas and Kuhn (1932) showed that their control pregnant rats given a complete diet that included fresh green vegetables and fruits, butter, yeast, and cod liver oil (a diet that was undoubtedly rich in magnesium, trace elements, and the B vitamins, as well as in vitamins A and D3 had uniformly successful gestations. To explore the influence of viosterol, calcium, and phosphorus, diets were prepared that lacked the above ingredients, and that provided adequate calcium and phosphorus and that were supplemented with or free of vitamin D2 (viosterol). Because of the absence of the additional nutrients in the "basic" experimental diet, the less successful gestations of the rats on that diet when viosterol was added reflects more than the influence of the vitamin D2 It is interesting, however, that the rats receiving the basic diet, adequate in calcium and phosphorus, did not tolerate the viosterol as well as did those on the diet that was deficient in calcium and phosphorus. When viosterol was given throughout pregnancy, none of the rats getting calcium and phosphorus delivered young; when given viosterol only during the last 14 days, two in ten rats came to term. One of the five calcium and phosphorus-deficient rats given viosterol throughout gestation came to term; five of seven given viosterol the last 10 to 14 days delivered young. Other than size and calcium and phosphorus ash content of the pups, no data were given as to their status at birth. The pups born to viosterol supplemented dams, whether or not they had had calcium or phosphorus deficiency, were larger and had higher calcium and phosphorus contents than did control pups or those on the basic diet.
Abnormalities in magnesium metabolism during pregnancy (as a result of, or a contributory factor in, vitamin D, PTH, CT, calcium, and phosphorus imbalances) have been shown to influence profoundly the success of gestation and the status of the newborn infant. Forfar (1976) has listed some of the mechanisms that can con tribute to disturbances in mineral metabolism in the perinatal period. He cited:
1. Inherent (genetic) defects in the parents transmitted to the offspring.
2. Congenital absence or hypoplasia of the parathyroids.
3. Disturbance of the maternal (intrauterine) mineral status with reciprocal fetal disturbances.
4. Nutritional deficiency.
5. Placental insufficiency and IUGR.
6. Prematurity.
7. Perinatal asphyxia and birth injury.
8. Excess phosphorus in infant feedings.
9. Eclampsia.
This listing is useful as a summation of many factors that have been presented in this section, interactions among which are frequent. Some additional data in several of the categories might further explicate some of the interrelationships, and shed some light on metabolic aberrations that might be contributory to clinicopathologic findings in the perinatal period.
3.5.1. Genetic Hypoparathyroidism
Absence or hypoplasia of the parathyroids is usually characterized by symptoms and signs of hypocalcemia. Although often also present, hypomagnesemia is less frequently sought and detected. This disorder is often associated with other endocrinologic abnormalities, including that of the thymus, and by lymphopenia and other immunologic deficiencies. This constellation of abnormalities is suggestive that magnesium deficiency might be an underlying factor, since it causes not only parathyroid dysfunction (Review: Nusynowitz et al. 1976) but also has been implicated in thymic hyperplasic and immunologic abnormalities (Reviews: Hass et al. 1976/1980; Larvor 1976/1980).
3.5.2. Genetic Hyperparathyroidism
Pregnant women with hyperparathyroidism generally have infants with at least transitory hypoparathyroidism (supra vide). However, familial hyperparathyroidism has been implicated in infants with laboratory or autopsy evidence of hyperparathyroidism. Hillman et al. (1964) reported two siblings with marked parathyroid hyperplasia, who were born to consanguinous parents. The first was detected at autopsy, in association with metastatic calcification and osteoporosis. The second was verified at surgery for subtotal parathyroidectomy. Goldbloom et al. (1972) encountered a second pair of siblings with hyperparathyroidism, with all of the typical characteristics: bone demineralization (with signs of rickets), elevated serum calcium and magnesium, and hypophosphatemia. Both survived subtotal parathyroidectomy: the first at 30 months of age, and the second after the first week of life. Their literature review uncovered nine additional cases in seven families. Not included in their list were two infants who had hypocalcemia and hyperphosphatemia, such as are seen with infantile hypoparathyroidism, but who were found to have parathyroid hypertrophy at autopsy (D. H. Andersen and Schlesinger, 1942). Since these infants had arterial calcification, no early rickets but osteitis fibrosis at the time of death at four months, it is possible that their pseudohypoparathyroidism might have been secondary to magnesium deficiency. The data from the infant reported by Monteleone et al. (1975) supports this speculation. He developed seizures on the ninth day of life and had slightly low serum calcium (7 mg/100 ml) and hyperphosphatemia. After intravenous calcium (which only partially controlled the seizures) his serum magnesium level was 0.8 mg/100 ml. Treatment with parenteral magnesium was more effective. A blood specimen taken three days later was found to have elevated iPTH levels. Thus, pseudohypoparathyroidism is another abnormality that might be related to magnesium depletion.
3.5.3. Reciprocal Maternal and Fetal Mineral Status
The maternal and intrauterine magnesium and calcium status has been considered in this section, as influenced by PTH, CT, vitamin D, and by impaired placental function, as well as in Chapter 2. The special role of excessive phosphate and vitamin D during infancy is considered in Section 4.3. The risk of prenatal vitamin D deficiency, as a cause of neonatal rickets, persists in groups with high vitamin D requirements. Whether the vitamin D refractoriness of magnesium deficiency might prove germane to the problem in pregnancy requires investigation.
3.5.4. Maternal Age and Parity: Diabetes Mellitus
It has been pointed out that very young (adolescent) mothers-who constitute many of the primiparous mothers with complications of pregnancy and premature or low-birth-weight infants-are at particular risk of poor dietary intake, including magnesium, the average intake of which is low during pregnancy. Young multiparous mothers, particularly those whose pregnancies have been frequent, and mothers of twins or greater multiple births, are also especially prone to magnesium depletion. Mothers with diabetes mellitus (a condition noted to be associated with hypomagnesemia even in the absence of pregnancy) have also delivered infants with subnormal magnesium levels. It has also been found that mothers of infants with neonatal hypocalcemic convulsions (such as have been shown to be associated with hypomagnesemia) are often significantly older, of higher parity, and of lower social class than controls (S. Roberts et al., 1973). Such mothers might be presumed to have been on suboptimal magnesium intakes, and to have been depleting their own magnesium stores with each successive pregnancy.
3.5.5. Eclampsia
Of particular importance are the low magnesium levels and high percentage retentions of pharmacologic doses of magnesium given to preeclamptic and eclamptic women. As has been discussed, the high fetal mortality of infants of eclamptic women is being increasingly attributed to placental damage, with resultant intrauterine malnutrition and hypoxia. Brash (1949) reviewed the literature to that time and evaluated 120 full-term live-born infants of toxemic mothers as compared with the same number of infants born after normal pregnancies. The incidence of abnormal lethargy, sometimes with edema or convulsions for days after birth was 11:1 in infants of toxemic mothers versus those born of normal mothers. Stillbirths and neonatal deaths occurred in 10.7 and 5.2%, respectively, of the infants born after eclampsia, and in 3.9 and 2.9% of those born after normal pregnancies. The observation that fetal salvage is improved in eclamptic women treated with magnesium sulfate alone, as compared with that of those given other antihypertensive and anticonvulsant medications (Zuspan and Ward, 1965; Zuspan, 1969) is further suggestive evidence of the importance of magnesium for both mother and infant.
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}