MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
Part III: Chapter 14
SKELETAL AND RENAL EFFECTS OF MAGNESIUM DEFICIENCY
14
Intensification of Magnesium Deficiency by Calcemic and Phosphate
Therapy
The possibility that magnesium deficiency might be contributory to, or might accompany, the abnormalities that cause osteopenia, hypocalcemia, hypercalcemia, and renal and cardiovascular disease is rarely considered in initiating therapy. Refractoriness to direct attempts to correct hypocalcemia and hypokalemia are now increasingly leading to investigation of serum magnesium levels, and less frequently to other (better) means of ascertaining the body's magnesium status. Emphasis is placed, in this chapter, on the problems that can result from treatment of either hypo- or hypercalcemia by agents that cause magnesium loss, when the primary disorder is one resulting in magnesium deficiency. Accepting the difficulties in evaluating the magnesium status, it is proposed that serum magnesium levels and 24-hour urinary magnesium outputs be made part of the routine initial diagnostic program. Since higher serum levels of magnesium are tolerable without serious hazard, except perhaps when there is hypercalcemia, it is suggested that magnesium therapy be tried before calcium loading of patients who have disorders that might make them susceptible to magnesium deficiency.
14.1. Calcemic Therapy during Pregnancy
It is usually recommended that pregnant women drink ample amounts of milk (which in most industrialized countries is "fortified" with antirachitic amounts of vitamin D) and take vitamin supplements that also provide an antirachitic dose of vitamin D. As pointed out earlier, the magnesium intake is likely to be meager. Then, when leg cramps of pregnancy develop (which can be caused by magnesium deficiency and hypomagnesemic hypocalcemia), the usual therapeutic approach is generally administration of calcium. Rarely is the magnesium status investigated, and magnesium treatment tried. There have been publications, however, that have shown that, both in normal and abnormal pregnancy, serum levels of magnesium tend to be low, even when corrected for hemodilution. Metabolic balance studies have shown that normal pregnant women should ingest sufficient magnesium to maintain a strongly positive balance, to meet both her needs and those of the fetus. It has been proposed that some of the abnormalities of pregnancy might be a consequence of magnesium deficiency. The fetus might be at even greater risk, experimental gestational magnesium deprivation causing greater fetal than maternal magnesium deficiency. Factors that increase magnesium requirements, such as gestational hypervitaminosis D, have caused congenital cardiovascular, renal, and skeletal defects. Suggestive evidence has been presented, and a theory promulgated that magnesium deficiency during pregnancy might be contributory to several "congenital" abnormalities of the heart, arteries, kidneys, and bones. Cardiac outflow abnormalities (such as can be produced by experimental hypervitaminosis D) have been found in conjunction with endocardial fibroelastosis. Infantile coronary or generalized arteriosclerosis, cardiomyopathy, and dysrhythmias, sometimes leading to sudden death, are seen alone or in combination with gross cardiac abnormalities. Such infants often have renal calcinosis, and if they survive the early months often have growth and mental retardation. Osteogenesis imperfecta, which resembles lesions that have been produced in pups of vitamin-D-poisoned rats, also resembles lesions of severe congenital hypophosphatasia, and is sometimes accompanied by cardiac and renal abnormalities, such as are seen in hypervitaminosis D. Neonatal hypoparathyroidism is common, and must be attributable to influences in utero, speculated to be gestational magnesium deficiency. One may wonder whether more intense magnesium depletion might so suppress the parathyroids in utero as to be responsible for congenitally deficient parathyroid tissue: "idiopathic" primary hypoparathyroidism. When the diseases are severe or familial, the patient's and the mother's intestinal absorption and renal tubular reabsorption of magnesium should be explored, as should that of other close relatives; since familial defects of magnesium metabolism have been recognized, it is conceivable that this might be a flaw that intensifies lesions caused by other heritable disorders, or even underlies some of them.
Them is a wide spread of vitamin D requirements and susceptibility to its toxicity. Thus, the practice of routinely providing much more than prophylactic amounts of vitamin D during pregnancy should be reevaluated, taking into account the usually high dietary intakes of phosphate, which, like low-magnesium intakes, intensifies vitamin D toxicity. Requiring investigation is the influence of such imbalances on the maternal organism and the placenta, and systematic investigation of the fetal organs and bones should be undertaken of stillborn infants, and of experimental models. Until definitive experimental data are available, we should keep in mind that magnesium has protected against experimental vitamin D and phosphate toxicity, and that magnesium deficiency (as is likely during pregnancy, especially in immature mothers and in women who have had frequent pregnancies, but also in less stressed mothers) has intensified the lesions of hypervitaminosis D and phosphate loads. Thus, magnesium supplementation (to a total of at least 7-10 mg/kg/ day) is suggested as a minimum for those without magnesium malabsorption or renal wastage. If either of those abnormalities of magnesium metabolism is detected 'and it should be sought if there is a familial history of suspect abnormalities), the magnesium supplementation should be correspondingly higher, and might have to be parenterally given.
14.2. Calcemic Therapy during Infancy
Considered in detail are the risks of treating neonatal hypocalcemia, which might well be a consequence of magnesium depletion, with calcemic agents. Failure of response of neuromuscular irritability is often the first clue to the necessity of evaluating the magnesium status, and favorable response to magnesium therapy the proof. However, during the time that a magnesium-deficient infant is being loaded with calcium, or given agents that cause bone resorption, damage can be inflicted on the heart, arteries, kidneys, and bones, and (especially in babies with genetic susceptibility to abnormalities of these tissues) permanent lesions might result. For example, magnesium deficiency and vitamin D excess each causes lipid abnormalities and damage to small and large arteries, respectively. In addition, the early renal lesions of magnesium deficiency (in the face of calcemic factors) are in the tubules, in the area of active magnesium reabsorption. Thus, such therapy might be contributory to establishment of transitory or permanent renal magnesium wasting. With continued calcemic treatment (or dietary custom that provides only moderate excess of vitamin D to infants who are hyperreactive to vitamin D, are magnesium deficient, or both), cardiac outflow abnormalities, endocardial fibroelastosis, premature atherosclerosis, renal calcinosis, and osteosclerosis as well as mental retardation, might result.
14.3. Calcemic Therapy for Osteopenias
The use of high-dosage vitamin D or its derivatives in the treatment of refractory osteopenias might similarly result in cardiovascular and renal damage, other soft tissue calcinosis, and osteosclerosis, rather than normal bone, which requires optimal magnesium for normal osteocyte activity and matrix formation. Little has yet been done to correlate the osteopenia or brittle chalky bones produced by either experimental magnesium deficiency or by vitamin D excess, the degree depending on the amount of calcium and phosphate in the diet. As regards the use of high-dosage calcemic agents for postmenopausal osteoporosis, reference should be made to the estrogen/parathyroid/magnesium interrelationships that suggest that magnesium's effect on osteocytes and matrix formation might find applicability in preventing further loss, if not serving to increase formation of organic matrix.
Inadvertent proof was provided that hypervitaminosis D produces metastatic calcification when very high doses of vitamin D were used to treat arthritis, even when the intake of calcium was not high (Danowski et al., 1945; Mulligan, 1947; Frost et al., 1947; Howard and Meyer, 1948; Reed, 1950; Christensen et al., 1951; Verner et al., 1958). In such instances, the calcium, phosphate, and matrix were drawn from the skeleton and deposited in soft tissues. In one of the studies (Frost et al., 1947) magnesium was studied and found to be low during the vitamin-D-toxic period and to rise when the overdosage was stopped. The evidence that some arthritic processes might be consequences of magnesium depletion suggests that seeking and correcting magnesium deficiency might be useful.
It is advisable to explore the magnesium status of patients with osteopenias before loading them with calcemic agents, which might prove useless in some or unduly toxic in others if magnesium deficiency is present. If hypercalcemia has already been induced by high doses of such agents as vitamin D or its congeners or metabolites, or by parenteral loads of calcium, the magnesium serum level and 24- hour urinary output should be determined. A parenteral magnesium load may be inadvisable until the hypercalcemia is corrected, and not by phosphate loading.
14.4. Treatment for Hypercalcemia
Because hypercalcemic crises are life-threatening, emergency treatment is directed toward lowering the circulating calcium levels quickly, by hydration with saline or dextrose in water, and increasing its urinary excretion with a potent diuretic such as furosemide, by administration of phosphate to increase its precipitation, hopefully in the bones, and by agents such as calcitonin to shift the calcium to bone, or mithramycin to antagonize bone resorption (Newmark and Himathongkam, 1974). Corticosteroids, which act more slowly, are recommended in long-term control of chronic hypercalcemia. Unfortunately, saline and furosemide diuresis, phosphate loads, and corticosteroids all increase magnesium loss, which is also caused by the hypercalcemia as well as frequently by the diseases that caused the hypercalcemia in the first place. Furthermore, inorganic phosphates have resulted in ectopic, sometimes fatal calcification (infra vide).
Hydration and furosemide diuresis are acceptable, until calcitonin can be obtained. Calcitonin is a preferable agent because it increases deposition of calcium in bone, stimulating bone alkaline pyrophosphatase (Orimo et al., 1970), without transferring calcium to soft tissue sites (Chausmer et al., 1965). In fact, there have even been reports that calcitonin protects against soft tissue calcification (Gudmundsson et al., 1966; Kenny and Heiskell, 1965; Gabbiani et al., 1968; Rasmussen and Tenenhouse, 1967; Rayssiguier and Larvor, 1974a). Once the plasma calcium levels are lowered, magnesium therapy can be substituted for the calcitonin, evidence having been obtained that calcitonin secretion is stimulated by increased magnesium (Radde et al., 1970; Bell and Kimble, 1970; Care et al., 1971; Littledike, 1970; Littledike and Arnaud, 1971; S. P. Nielsen, 1974). Additionally, moderately increased magnesium levels suppress parathyroid secretion (Care et al., 1966; Buckle et al., 1968; Gitelman et al., 1968a; Massry et al., 1970b; Sherwood, 1970; Sherwood et al., 1970; Altenahr and Leonhardt, 1972). Competition between calcium and magnesium for a common renal tubular reabsorptive pathway (Samiy et al., 1960a,b; Charbon and Hoekstra, 1962; Ardill et al., 1962; Heaton et al., 1964; Massry and Coburn, 1973) has also been credited for the increased urinary excretion of calcium and drops in serum calcium that accompany magnesium loads (Womersley, 1956; Chesley and Tepper, 1958; Kelly et al., 1960; Kemeny et al., 1961: S. P. Nielsen, 1970; Nielsen and Jorgensen, 1972).
It is recommended that magnesium not be given until the acute hypercalcemia been lowered, intensification of soft-tissue calcinosis having been produced by magnesium given to rats with experimental hypercalcemia caused by hypervitaminosis D (Whittier and Freeman, 1971).
14.4.1 Risks of Phosphate Therapy
Inorganic phosphate therapy has been utilized and warned against for many years in the treatment of hypercalcemia and of skeletorenal disorders. Oral administration of inorganic phosphates was found, almost 50 years ago, to reduce the acute hypercalcemia of patients with hyperparathyroidism (Bulger et al., 1930; Albright et al., 1932). However, both groups of investigators expressed concern about the risk of promoting nephrolithiasis or other extraskeletal calcification. Bulger et al. (1930), for example, found extensive calcification of lungs, gastric mucosa, and kidneys when a patient died of bronchopneumonia a few days after the infusion. Shortly thereafter, Bulger and Gausman (1933) demonstrated that hyperparathyroidism causes negative magnesium balance. In 1962, Dent reintroduced phosphate therapy for hypercalcemia. One of his patients responded well; the other developed extensive painful ectopic calcification. Four years later, R. S. Goldsmith and Ingbar (1966) again described the usefulness of phosphate loads for treatment of life-threatening hypercalcemia, applying it also to patients with neoplasms. They obtained rapid and dramatic decreases of serum calcium levels and improvement of symptoms in 16 of their 20 patients. Ten died, of whom 7 had autopsies. Five had extraskeletal calcification. One, who had not been examined postmortem, had died of a massive infarction the day after the phosphate infusion. Because of uncertainty that these six instances were related to the treatment, and of the rapidity with which the phosphate lowered the plasma calcium level, R. S. Goldsmith (1970) reiterated his recommendation that this approach was most va1uable for hypercalcemic crisis in his critical review of Eisenberg's (1970) caution as to the risk of producing metastatic calcification. Eisenberg (1970) noted the instances in which such calcification had been reported after either intravenous or oral administration of large doses of phosphates, and warned of the likelihood that the calcium would precipitate out in soft tissues. For example, Schackney and Hasson (1967) reported hypotension and acute renal failure in two patients whose hypercalcemia had been treated by phosphate infusions. One had extensive metastatic calcification in the heart, lungs, kidneys, and pancreas; the other exhibited no metastatic calcification on autopsy. Breuer and LeBauer (1967) reported a patient with multiple myeloma and hypercalcemia, who had a good temporary clinical and chemical response to intravenous and oral phosphate treatment, but who suddenly died with renal insufficiency and pneumonia and was found to have extensive pulmonary and renal calcification. Carey et al. (1968) reported metastatic calcification involving the endocardium, coronary arteries, and kidneys (glomerular, intraluminal, and interstitial) in a patient whose hypercalcemia of neoplastic origin had been treated with inorganic phosphate infusions. Marti and Cox (1970) reported additional patients who developed irreversible calcinosis, particularly of renal tubules and lungs, following phosphate infusions for hypercalcemia resulting from bone metastases. Dudley and Blackburn (1970) recommended slit lamp conjunctival examination to detect early extraskeletal calcification, such as they found in seven of nine patients who had been treated with high-dosage oral phosphates. Five had been treated for hypercalcemia; two with hyperparathyroidism developed impaired renal function during therapy. Of four normocalcemic patients, who were being given phosphate therapy for renal calculi, three developed conjunctival calcification, one developed radiologic evidence of renal and one of arterial calcification.
Thus, although inorganic phosphate has been effective in reducing hypercalcemia and the incidence of calcific urinary stones, it carries the risk of soft-tissue calcinosis, such as is seen with magnesium deficiency, and is intensified by phosphate loading. Monsaingeon et al. (1971/1973) found that oral inorganic phosphate loads (2.25 g/day) decreased the urinary magnesium concentration more than it did that of calcium in 70% of 29 patients with urinary calculi. They cautioned that it is necessary to monitor magnesium levels in patients treated with phosphates.
Short-term administration of cellulose phosphate to normal subjects has reduced the intestinal absorption and urinary output of both calcium and magnesium (Dent et al., 1964), and caused a gradual decline in serum magnesium (but not calcium) levels (Parfitt, 1975). That long-term administration of cellulose phosphate, given to reduce the urinary calcium in stone formers, can cause magnesium depletion is indicated by Sutton's (1968) study. He reported hypomagnesiuria and hypomagnesemia in a recurrent stone former, who had been treated with a low calcium diet and oral cellulose phosphate for 6 years. His plasma magnesium remained between 0.65 and 1.25 mEq/liter and his 24-hour urinary magnesium between 2 and 15 mg during a year of observation, while he was on that regimen.
Other phosphates have induced fewer problems, possibly in part because of lesser depletion of magnesium, and in part because of their increase in urinary output of inhibitors of calcium crystallization in urine. For example, orthophosphate (disodium hydrogen phosphate dihydrate) administration (12 g/day = 1.98 g P/day) to patients with recurrent renal calculi, to reduce the intestinal absorption of calcium and thereby to reduce urinary calcium excretion, was found also to increase the urine citrate and pyrophosphate levels, but to influence the magnesium levels and balance only slightly. It caused a more profoundly negative calcium balance, decreased urinary calcium output, but caused a net increase in the Ca/Mg urinary ratio. The crystallizing propensity was reduced, probably largely because of the orthophosphate-induced pyrophosphate and citrate levels. The investigators who showed that inorganic pyrophosphate inhibits the precipitation of hydroxyapatite crystals in vitro (Fleisch and Neuman, 1961) developed condensed phosphates (diphosphonates) that are less readily hydrolyzed and that are effective in preventing ectopic calcification in vivo (Irving et al., 1966; Francis et al., 1969). A diphosphonate, which is under investigation for its inhibition of ectopic calcification and of bone resorption (Francis et al., 1969; Fleisch et al., 1969; Russell et al., 1971; Michael et al., 1971; Saville and Heaney, 1972), has been shown to cause negative magnesium balance on long-term in children with ectopic calcification (Uttley et al., 1975).
With so much evidence that magnesium deficiency accompanies hypercalcemia and its treatment, it is tempting to recommend prompt magnesium repletion. However, one must keep in mind the magnesium dependence of phosphatases that destroy the polyphosphates (including the pyrophosphates) that inhibit precipitation of calcium salts in the soft tissues. In fact, several of the British investigators, who reported the severe form of infantile hypercalcemia, suspected that use of magnesium laxatives might have intensified the syndrome that is characterized by renal, cardiovascular, and brain damage and calcinosis. Thus, as indicated earlier, serum calcium levels should be lowered first, by the least dangerous means, before instituting magnesium therapy. Without hypercalcemia or hyperphosphatemia, magnesium activation of soft-tissue alkaline or pyrophosphatase should not present a danger of precipitation of calcium phosphate salts. Magnesium stimulation of bone alkaline or pyrophosphatase should function to take up calcium and phosphate, particularly if the magnesium suppresses parathyroid and increases calcitonin secretion.
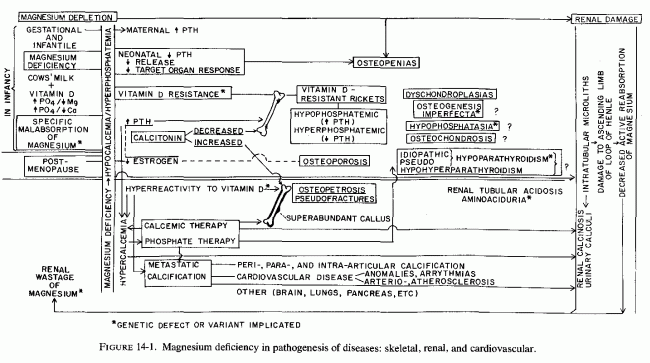
Among the tissues damaged by magnesium deficiency, those of the cardiovascular and skeletal and the urinary tract are listed on Table 14-1. Some of the abnormalities cited in several of the diseases to which there is reason to believe magnesium deficiency is contributory are disorders that are comparable to those seen in experimental magnesium deficiency. Unfortunately, some of the early findings (such as hypocalcemia, neuromuscular irritability, and osteopenias) suggest direct treatment with the obviously deficient substance, calcium, or by agents that normally function to increase calcium absorption and its blood levels. When the hypocalcemia or osteopenia is secondary to magnesium depletion, such treatment can intensify the magnesium loss, increase the cellular damage (caused by magnesium deficiency), and lead to metastatic calcinosis (Figure 14-1).
Thus, the underlying abnormality (metabolic or dietary or both) that prevents normal magnesium utilization can lead to abnormal function or response to parathyroid hormone, vitamin D, or calcitonin, with possible production of a variety of osteopenias. Most of the diseases have their roots during gestation or early infancy. One, postmenopausal osteoporosis, is entered because of the possibility that the drop in estrogen secretion might contribute to relative hyperparathyroidism. High dietary phosphate intakes, which can contribute to major disorders of infancy, might also play a significant role in the high incidence of osteoporosis and periodontosis later in life. This can be intensified in the treatment of life-threatening hypercalcemia of malignant disease and hyperparathyroidism.
The use, not only of phosphate therapy (which has a distinct risk of metastatic calcinosis), but of high-dosage calcemic agents during infancy (to counter hypocalcemia and refractory osteopenias), and to treat postmenopausal, senile, or disuse osteoporosis, also intensifies magnesium loss and metastatic calcification. Such treatment also increases bone mineralization, but in the absence of optimal magnesium, the bone has abnormal matrix. Such treatment is likely to cause increased bone density, but decreased bone elasticity, with resultant marblelike, brittle bones. Renal dysfunctions-tubular acidosis, aminoaciduria, and calcinosis and calculi- might also result from magnesium deficiency, intensified by calcemic therapy.
This book presents evidence that early investigation of the magnesium status is important. Whether use of magnesium supplements during gestation and infancy will reduce the incidence of some of the indicated congenital anomalies will require many years to ascertain. Clues might be obtained from experimental models, prepared so as to mimic some of the nutritional imbalances, and to exaggerate magnesium deficiency, such as might be found with genetic magnesium malabsorption or renal wastage. Since magnesium administration is benign (unless there is renal failure), it is proposed that prophylactic and therapeutic trials are justifiable.
This particularly true for patients with premature cardiovascular disease, or for subjects with familial histories suggesting high risk of early ischemic heart disease or strokes. It is also true for patients with the bone disorders cited, and for those with functional renal disorders such as tubular acidosis and aminoaciduria, and for patients with calcific urinary tract disease.
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
{kind=link}
{kind=link}