MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
Part III: Chapter 12
SKELETAL AND RENAL EFFECTS OF MAGNESIUM DEFICIENCY
12
Abnormal Bone in Magnesium Deficiency
Bone-wasting diseases that are resistant to physiologic doses of vitamin D, calcium, or phosphate and that have been treated with pharmacologic doses of each or of combinations of mineralizing agents, are likely to be associated with magnesium deficiency. In some instances, initial magnesium inadequacy might be contributory to the osteopenia, as in hyperparathyroidism, secondary to malabsorption, with hemodialysis with low-magnesium water, or possibly in pregnancy. There is suggestive evidence that severe magnesium depletion (in utero), alone or with hypervitaminosis D, might participate in abnormal fetal bone formation that might find expression as fractures of low-birth-weight infants, osteogenesis imperfecta, or hypophosphatasia. In infancy and later in life, vitamin-D- or parathyroid-refractory osteomalacia or hypocalcemia might also have magnesium depletion as a contributory factor. Failure to correct the magnesium deficiency before use of calcemic therapy has failed to correct hypocalcemia. In those with osteopenia (to which magnesium deficiency has contributed), failure to correct that deficit before starting aggressive mineralizing therapy intensifies the imbalance.
Apart from the risk of thereby increasing the risk of extraskeletal damage and calcinosis, such treatment can adversely influence the skeletal system. It can lead to hypermineralization of bone or abnormal matrix, in some instances with exuberant osteoid formation. Marbleized or chalky brittle bones (such as have been described in rats on diets rich in calcium, phosphate, and vitamin D, and poor in magnesium) might develop. If the diet is high in phosphate but poor in calcium and magnesium, osteopenia has been seen. Low magnesium intakes, when severe, have been associated with desmoidlike tumors; whether the abnormal osteoid at sites of pseudofractures is a disorder of common etiology remains to be determined. It is provocative that severe magnesium depletion is most commonly recognized with vitamin-D- or PTH-refractory hypocalcemia, usually without bone wasting.
12.1. Osteopenia of Magnesium Deficiency (Animals)
It has long been known that magnesium-deficient animals have arrested growth (Leroy, 1926; Kruse et al., 1932), but the precise nature of the bone abnormalities produced has not received much attention. In 1930, Huffman et al. reported that the ribs of magnesium-deficient calves are easily broken at the sternal ends, and that the specific gravity of the long bones is subnormal. Cunningham (1933) reported that rats on diets low in magnesium have narrowed epiphyseal plates, which contain few chondrocytes, and that the subepiphyseal region has few trabeculae. Duckworth et al. (1940) noted the fragility of the long bones of their magnesium-deficient rats and attributed it to abnormal matrix. Yamane and Singer (1953) observed that metaphyseal bony trabeculae are lost and that the zone of preliminary calcification just below the epiphysis is atrophic or absent in magnesium-deficient hamsters. Blaxter (1956), who found no histological evidence of abnormality of the calcification process in bones of acutely magnesium deficient calves, speculated that the matrix might be adversely affected. E. R. Morris and O'Dell (1961) postulated that magnesium deficiency interferes with cellular function of hard tissues, thereby preventing formation of normal matrix.
Since then, firm evidence has been obtained that magnesium deficiency does, in fact, interfere with normal formation of the matrix of both bones and teeth. Bernick and Hungerford (1965) found differences in staining characteristics that suggested that the ground substance of the magnesium-deficient matrix contains mucopolysaccharides that are less polymerized and less subject to normal calcification. They also compared the histologic differences between the epiphyses of the rats fed a magnesium-deficient diet for 19 days, and those of controls. They confirmed the early evidence that the cartilage of the epiphyseal plate of the heads of the tibias of the deficient rats is slightly narrower than normal. They found trabeculae, extending from the metaphysis into the diaphysis, that are shorter and wider than normal. The proximal epiphyseal cartilages of the deficient rats exhibit a slight decrease in number of proliferating cells, and relative increase in the number of hypertrophied cells, and a decrease in the width of the calcifying matrix. Clark and Bélanger (1967) also showed thinner epiphyseal plates in magnesium-deficient rats, and few chondrocytes. There were practically no new trabeculae at the subepiphyseal area, and the diaphysis contained immature matrix with small elongated osteocytes. This was confirmed in later studies (Hunt and Bélanger, 1972; Bogoroch and Bélanger, 1975).
Trowbridge and Seltzer (1967) investigated the effect of acute magnesium deficiency on the organic matrix of bone and dentin employing the uptake and tritiated proline to assess collagen formation, 35SO4 to assess sulfation of protein polysaccharides, and measuring the biochemical localization of alkaline phosphatase in bones and dentin. They found minimal osteoblastic activity, with marked suppression of the amount of tritiated proline uptake in the collagen of the bone matrix in the deficient rats. Also, sulfation of glycosaminoglycuronoglycans was diminished in the osteogenic layer of the periodontal ligament, and reduced intensity of staining for alkaline phosphatase in the periodontal ligament and in osteoblasts suggests that magnesium deficiency decreases bone alkaline phosphatase; serum levels of the enzyme were also low. Hunt and Bélanger (1972) also showed that the cartilage matrix of magnesium-deficient rats appeared depleted in sulfated mucopolysaccharides. The underlying bone spicules of the epiphyseal plate of the tibia were thin and poorly ossified, as were the diaphyses.
Lai et al. (1975) have verified the reduction in bone magnesium, phosphatase, and matrix in magnesium-deficient rats and found hypermineralization with increased bone ash and calcium content. Their findings were more similar to those of Orent et al. (1934) and Watchorn and McCance (1937) than they were to those of Duckworth et al. (1940). The former two groups had fed their rats diets high in calcium and vitamin D; the latter used less calcium and did not indicate use of vitamin D. [Lai et al. (1975) did not specify the nature of the basic diet, but basic rat diets, provided currently from most firms, are rich in calcium, phosphorus, and vitamin D.] The bones were more brittle than those of control rats.
Bone matrix implanted in skeletal muscle of magnesium-deficient rats, and controls fed the same commercially supplied deficient diets, but supplemented with 265 mg/100 g of diet showed marked differences in bone growth and development (Belanger and Robichon, 1975). There was osteoporosis in the lumbar vertebrae of the magnesium-deficient rats, and very little bone formed on either the inside or the outside of the implants. Only 2 of the 15 rats that survived 3 weeks formed new trabeculae, and these were fibrillar and poorly mineralized. At several sites, there were small amounts of bone tissue mixed with islands of chondrocytes, surrounded by precartilaginous or poorly differentiated matrix. Further outward from the implant there was an "envelope" of fibroblastlike cells and collagen that separated the implant from the muscle. In half of the specimens, there was invasion of the implant by thin cartilage wedges, coming mostly from the periphery of the implant. Near these wedges, there was deterioration of the implant matrix. In contrast, the magnesium-supplemented rats had formed well-mineralized trabecular bone inside and outside the implant. In a few areas, cartilage or a mixture of cartilage and bone had formed.
12.2. Abnormal Bone: Hypermineralization and Hyperplasia of Magnesium Deficiency
Paradoxically, magnesium-deficient animals have exhibited excess (abnormal) bone growth in addition to osteopenia. Generalized medullary bone growth (osteomyosclerosis and periosteal tumors of the desmoid type occurred at the femoral linea asperea in severely magnesium-deficient rats (Bélanger and Hunt, 1971/1973; Hunt and Bélanger, 1972). (This is a prominent longitudinal ridge on the middle third of the bone, with rich vascular supply, and concomitant sites of accretion and osteocytic osteolysis, which indicate that it is an area of considerable metabolic activity.) The degree of magnesium deficiency was critical in the formation of both the periosteal tumors and intramedullary bone; both abnormalities were produced only in severely magnesium-deficient rats, but irrespective of the concentration of bone magnesium. The investigators speculated that matrix and bone cells could be differentially depleted of magnesium, and that the bony overgrowth was related to changes in the magnesium concentration in the organic phase of bone. The periosteal tumors rapidly disappeared when the rats were supplemented with magnesium. This was interpreted as indicative of magnesium depletion-induced accumulation of cells unable to differentiate properly, possibly as a result of enzymic malfunction. Deficient rats that developed fibrous hyperplasia showed high concentration of alkaline phosphatase activity throughout the hyperplastic membrane (Bélanger et al., 1972a).
Although the periosteal desmoid tumor was first shown to be a characteristic of magnesium deficiency by this group (Hunt and Bélanger, 1972), the authors noted that Duckworth et al. (1940) had referred to "disordered growth of the organic matrix of leg bones" in their magnesium-deficient rats that might have been a comparable phenomenon. Lai et al. (1975) later observed that 10 of their 11 magnesium- deficient rats had tumorlike femoral exostoses.
These tumorlike growths resemble that described by McCance (1946) in an adolescent girl who developed weakness and hypophosphatemic vitamin-D-resistant osteomalacia at the age of 15. She had multiple spontaneous pseudofractures and callus formation of her long bones (Looser's nodes), and had a tumor on the shaft of the tumor. Histologic examination showed abnormal osteoid tissue that was not considered neoplastic. Metabolic balance studies, done while the patient was receiving about 2000 units of vitamin D daily, showed substantially negative balances of calcium, phosphorus, and magnesium. On a daily magnesium intake of about 230 mg, she lost an average of 25 mg/day over a 7-day period. Massive vitamin D therapy (500,000 units/day) greatly improved her retention of calcium and phosphorus but improved the magnesium retention only slightly. The vitamin D was stopped when signs of toxicity developed (after a month), and her magnesium retention improved markedly (to + 40 mg/day). When her magnesium intake was increased (to 390 mg/day) she went into strongly positive magnesium balance (+ 90 mg/day) and showed steady clinical improvement.
It is provocative that similar exostoses, described as irregular subperiosteal new bone formation or exuberant callus, have been reported in patients with hypophosphatasia (Schlesinger et al., 1955; Currarino et al., 1957), a condition postulated to be related to magnesium depletion. Hypophosphatemic rickets has also been associated with profound weakness and Looser's nodes in an adult, who also had a lengthened QT interval (Milne et al., 1952). The authors attributed the weakness and abnormal ECG to her hyperkalemia. The bone and cardiac manifestations might also have had magnesium deficiency as a common cause.
Hunt and Bélanger (1972) found that parathyroid activity influenced the nature of the bone tumor produced by experimental magnesium deficiency. Parathyroidectomized magnesium-deficient rats had a large tumoral mass that consisted of layers of fibrous tissue on the outside, then cartilage, and an internal layer of bone. Administration of PTH to these animals reduced the amount of cartilage, which then appeared as small peripheral isolated units, and resulted in abundant growth of medullary bone throughout the central cavity of the femur and tibia. In view of the estrogen/PTH antagonism in bone accretion and resorption (Ranny, 1959; Review: Seelig and Lehr, 1971/1973), the observation that ovariectomy, with and without estradiol administration, modified the incidence and severity of skeletal lesions caused by magnesium deficiency (Bogoroch and Bélanger, 1975) is provocative. Although the reduction in diaphyseal width of magnesium-deficient ovariectomized rats did not differ significantly from that seen in intact deficient rats, some of the ovariectomized rats showed greater subperiosteal hyperplasia. Estradiol-treated magnesium-deficient rats had a quarter the incidence of subperiosteal hyperplasia seen in untreated magnesium-deficient rats. Possibly related is the fourfold increase in plasma alkaline phosphatase activity produced by estradiol treatment of chickens (Malinow et al., 1960), since alkaline phosphatase synthesis and activity are magnesium dependent and the enzyme is involved in bone metabolism.
Bone tumors have also been experimentally produced by beryllium (Janes and McCall, 1975), which inhibits alkaline phosphatase, an inhibition that is partially reversed by adding magnesium (Aldridge, 1950; Grier et al., 1949). It is thus of interest that both in human and experimental osteogenic sarcoma, magnesium bone tumor levels were low (Janes et al., 1972; Jones and McCall, 1975). This brings us to the discussion by Hunt and Bélanger (1972) of the significance of their observation of the osteomyelosclerosis and subperiosteal tumors of their magnesium-deficient rats. They noted that the deficiency-induced lesion seemed to correspond to the periosteal desmoid described in the Catalogue of Tumors of Bone and Cartilage (Spjut et al., 1969), which is described as midway "between a true tumor and nontumorous connective tissue hyperplasia." They also noted that the occurrence of osteomyelosclerosis occurs in human disease, frequently in association with certain forms of leukemia and other blood dyscrasias and that leukemia has occurred in magnesium-deficient rats (McCreary et al., 1967; Battifora et al., 1968).
Thus, magnesium deficiency causes major metabolic disturbances of the bone that can lead to osteopenic yet hypermineralized brittle bones as well as hyperplasia and might even participate in an early neoplastic process. The degree of the deficiency as well as the concomitant dietary imbalance and hormonal responses, affects the nature of the lesion produced. Possibly much of its direct effect on bone is mediated by its effect on synthesis or activation of phosphatases, by its effects on bone matrix, and by its influence on differentiation of the bone cells.
12.3. Bone Diseases Possibly Related to Magnesium Deficiency
12.3.1. Fetal Magnesium Deficiency and Bone Damage
12.3.1.1. Interrelationships with Parathyroid Hormone and Calcitonin
The failure of maintenance of fetal magnesium levels at the expense of the mother can result in responses in PTH and CT secretion that can influence fetal bone growth and development. Both the parathyroids and the C cells are functional early in fetal life. They are influenced by the maternal magnesium and calcium levels, both of which have been shown to have a tendency to below. Both cations, unlike PTH and CT, can cross the placental barrier, and thus must be controlled by fetal parathyroid and C-cell responses. This homeostatic control is mediated by their effects on fetal bone. Fetal rat bone, kept in a medium low in magnesium (0.3 mM), showed less release of tagged calcium when exposed to PTH (Raisz and Niemann, 1969), an effect like that seen in the intact magnesium-deficient experimental animal or human. Fetal bones in media high in magnesium (4.3 mM) showed the same release of calcium from bone as did bones kept in physiologic magnesium and calcium concentrations when PTH was added. Bone resorptive activity of human fetal parathyroids has been demonstrated as early as 12 weeks gestation, at which time there are secretory granules (Scothorne, 1964). Differentiation of cellular organelles are manifest later in fetal life (Altenahr and Wohler, 1971). That fetal thyroid tissue can secrete CT early in gestation is suggested by the better growth of fetal chick bones in the presence of 8-day fetal chick thyroid tissue than in its absence (Gaillard and Thesingh, 1968). There is increased fetal CT secretion in rats toward the end of gestation (Garel, 1970; Feinblatt and Raisz, 1971). Calcium infusion in the fetus has suppressed the hypocalcemic effect of CT (Garel, 1970). It has been suggested that this effect might be mediated by inhibition of bone resorption, as in the adult. Exposure of fetal rat thyroid tissue in vitro to increasing concentrations of calcium (to 2 1/2 times normal levels) increased the release of CT; it inhibited PTH stimulated calcium release from fetal bone threefold (Feinblatt and Raisz, 1971). When both calcium and magnesium concentrations were physiologic (Ca/Mg = 1.0/0.8 mM) additional CT caused some inhibition of PTH-stimulated release of 45Ca from fetal bone. When the magnesium concentration was reduced to 0.4 mM or raised to 1.6 mM a slight increase in CT secretion resulted, which increased the percentage inhibition of release of calcium slightly. When magnesium was further raised to 3.2 mM the CT-induced percentage inhibition of calcium release rose a little more, although none of the changes were sufficient to be considered significant. There has been considerable experimental evidence that CT increases bone calcification and new bone formation directly (Matthews et al., 1972; Wase et al., 1967; Pallasch, 1968; Ziegler and Delling, 1969; Delling et al., 1970; Gaillard and Thesingh, 1968; Salomon et al., 1973), independent of its counteraction of demineralization. Thus, the lowering of plasma levels of calcium, magnesium, and phosphorus in response to CT (Garel et al., 1968, 1969; Garel and Barlet, 1974) might indicate utilization of those elements in bone formation. The high CT levels in the fetus are likely to take part in bone growth and calcification (Samaan et al., 1973b).
12.3.1.2. Interrelationships with Gestational Hypervitaminosis D
Magnesium deficiency has been implicated in placental and fetal abnormalities. Placental calcification has been reported in magnesium-deficient rats (Dancis et al., 1971). Since most rat diets are rich in calcium, phosphorus, and vitamin D, further work is necessary to determine how much of the placental damage in that study might have been caused by the other nutrients. Hypervitaminosis D during pregnancy has been implicated in human and experimental placental damage. In rats, it has also caused fetal bone damage (Ornoy et al., 1969). That gestational hypervitaminosis D (which increases magnesium loss) causes both placental and fetal bone damage is provocative, but does not separate the possible direct effect of vitamin D toxicity from the presumed effect of magnesium deficiency on the placenta. That magnesium deficiency causes bone damage has been clearly demonstrated after birth. Vitamin D excess, given to pregnant rabbits, has caused premature closure of the fontanels, osteosclerosis, and palatal abnormalities (Friedman and Mills, 1969). Nonetheless, the levels of the 25-OH-D3 metabolite of young of rabbits given toxic doses of vitamin D have been subnormal (Mehlhorn et al., 1977), suggesting abnormality in vitamin D metabolism under these conditions.
Detailed study of the fetal bone abnormalities caused by toxic dosage of vitamin D in pregnant rats (40,000 units D2) showed that the fetuses had 61% decreased bone ash by the 21st day of gestation, shortened thin diaphyses, and abnormal epiphyseal cartilage. Pups of rats given half as much vitamin D (Ornoy et al., 1972) had bone deformities consisting of kyphoscoliosis and distortions of the long bones. There was less osteoid in the metaphyses, there were many metaphyseal fractures, and diaphyseal bone was short, distorted, and with much thinner than normal periosteal bone. By the 30th postnatal day, some of the pups had epiphyseal fractures. The authors observed that the prenatal vitamin D excess resulted in lasting defects in bone formation and imperfect healing of fractures in the newborn, that resembles some of the characteristics of osteogenesis imperfecta. When prenatal vitamin D excess causes osteopenia, it is possible that magnesium deficiency might complicate the picture, in that it might militate against CT release, high intakes of magnesium (like hypercalcemia) stimulating CT secretion. It should be kept in mind that fetal magnesium stores are likely to be suboptimal in magnesium-deficient mothers. Experimental magnesium deficiency has caused bone lesions (supra vide). Epiphyseal separation and osteochondrosis have been reported in premature infants (Griscom et al., 1971) and have been reported in older children with magnesium deficiency (Miller, 1944; Klingberg, 1970) or with the bone lesions of celiac disease of children (Parsons, 1927) or adults (Bronsky, 1970; Prost et al., 1972), which has been associated with either magnesium or vitamin D deficiency or both (Prost et al., 1972). It has also been associated with "pseudohypoparathyroidism" with parathyroid hyperplasia and hyper- rather than hypoparathyroidism, and is resistant to the action of PTH, further suggesting magnesium depletion.
12.3.2. Magnesium Deficiency and Bone Disease in Low-Birth-Weight Infants
Low-birth-weight infants, who might reflect intrauterine malnutrition rather than prematurity, are especially prone to magnesium depletion. The pathogenesis of the lesions described in this section is difficult to understand and interpret, conflicting findings having been reported and many factors interrelating. For example, magnesium depletion can cause decreased release of PTH and decreased target organ responsiveness (Fig. 12-1.). Yet acute magnesium deficiency has increased PTH release and chronic suboptimal magnesium intake has caused parathyroid hyperplasia (Larvor et al., 1964a). Vitamin D excess causes hypercalcemia, which can increase CT secretion and cause osteosclerosis (Fig. 12-2) or can cause magnesium loss and osteopenia (Fig. 12-1). Magnesium deficiency in infancy can cause hypocalcemic tetany and can be involved in vitamin D refractoriness. Both magnesium and vitamin D deficiency can cause fetal, neonatal, and later osteopenias. Yet mothers with presumptive magnesium deficiency and placental pathology have given birth to infants who developed osteosclerosis almost indistinguishable from that of rodents with hypervitaminosis D. Conversely, mothers with intestinal malabsorption, which probably interfered with absorption of both magnesium and vitamin D, gave birth to infants with congenital rickets. Furthermore, low-birth-weight infants have been shown to require vitamin D supplementation, above that in their fortified formula to avoid rickets (Lewin et al., 1971). It should be recalled, here, that magnesium deficiency increases vitamin D requirements. Vitamin D supplements have increased, significantly, plasma 25-OH-D3 levels in maternal, cord, and neonatal blood (Belton et al., 1975, 1977). The possibility of abnormal vitamin D utilization with gestational excess (Mehlhorn et al., 1977) should be kept in mind, low levels having been seen.
Experimental evidence has been presented that suboptimal supply of magnesium to the fetus can stimulate fetal PTH secretion. Whether it can contribute to abnormal vitamin D metabolism in the mother or the fetus should be explored. The degree to which maternal and fetal bone stores are utilized (under the influence of PTH secretion and vitamin D administration) probably depends upon the ratio of PTH and calcitonin (CT) levels and the metabolism of vitamin D, which are affected by both calcium and magnesium levels. Although fetal osteosclerosis has been correlated with excessive vitamin D, calcium, and phosphate administration, magnesium-deficient fetuses are also at risk of bone loss or defective formation.
Indirect evidence that this can be so derives from the osteoporosis, epiphyseal separation, poorly mineralized subperiosteal new bone, enlargement of costochondral junctions, metaphyseal cupping, and spontaneous fractures in three premature infants of women with placental pathology. Two of the mothers were preeclamptic and one was a young multipara who had two previous premature deliveries (Griscom et al., 1971). All of the women were probably magnesium deficient, two as a result of bearing twins and having preeclampsia, and one because of frequent pregnancies at a young age. That the infants might also have been magnesium deficient is suggested by the fact that two were survivors of twin pregnancies and one was premature. The twins who did not survive had been stillborn in one instance and had died at 10 hours in the other, the latter with thymic involution (such as is seen in infants with long-standing intrauterine distress) and with microfocal myocardial necrosis. The bone disease of these three infants was diagnosed a week before death: a few days after sudden cardiac arrest at 71 days in one, and two months after a cardiac murmur was diagnosed at one month in another. Additional suggestive evidence that magnesium deficiency might have been present was the severe anemia that developed in all three, such as has been produced in the young by experimental magnesium deficiency in pregnant rats (Cohlan et al., 1970) and in nonpregnant rats (Elin et al., 1971b; Elin, 1973, 1976/1980)
Griscom et al. (1971) pointed out that demineralization of bone may not be rare in low-birth-weight infants in the early weeks or months of life. The first such case, an atrophic newborn infant with osteogenesis imperfecta in association with arterio sclerosis, was reported by Johansson (1921-1922). Dystrophic osteomalacia of prematurity has been reported from France (Boissiere et al., 1964), and is characterized by icterus and pneumonitis as well as by bone disease. Griscom et al. (1971) found many similarities in the three infants they reported to those of the French infants (Boissiere et al., 1964). The disorder usually does not become manifest until the third month of life and commonly appears in twins. Fractures, subperiosteal new bone, and osteoporosis characterize the disease. However, only one of the three American infants of Griscom et al. had icterus, and that to only a slight degree. It was seen in 22 of 26 of the French infants. The American infants also did not present with hypocalcemic tetany, such as was reported from France. Griscom et al. (1971) considered the picture to reflect a metabolic, probably nutritional disorder other than rickets, and considered it likely to be fairly common among premature infants.
Another premature infant that developed rarefaction of ribs and scapula and spontaneous rib fractures by the third month of life, and also had anemia considered typical of prematurity, was diagnosed as rachitic (Keipert, 1970). This infant was the fourth child born in a difficult labor to an apparently normal mother. Intermittent apneic attacks began at 11 days. Despite vitamin D supplementation of 1,400-800 IU/d, some evidence of rickets persisted at nine months of age. The author commented that fractures are more common in rachitic than in normal bones, but observed that nonrachitic premature bones are also easily traumatized. He noted that the subperiosteal proliferation of prematurity is not related to vitamin D deficiency , and that Eek et al. (1975) found such changes earlier in premature infants fed cows' milk than in breast-fed prematures. Eek et al. (1957) postulated that double periosteal contours appeared in such infants when deposition of minerals increased after a period of poor mineralization. Tsang et al. (1977) have reviewed data on the abnormal and delayed skeletal mineralization in very low-birth-weight infants. Their group has shown that extrauterine bone mineralization lags significantly in such infants (Minten et al., 1976; Steichen et al., 1976).
It is provocative that the low-birth-weight infants who develop bone lesion rarely exhibit symptomatic hypocalcemia, such as is seen in those free of osteopenia. Possibly fetal hyperparathyroidism had mobilized fetal bone calcium. However, an alternative possibility must also be considered, that of the response of maternal, fetal, and neonatal C cells to changes in calcium and magnesium levels. High CT levels might contribute to both low plasma levels of calcium and magnesium, increasing bone mineralization.
12.4. Magnesium Status and Vitamin D Requirements and Responses
12.4.1. Increased Vitamin D Requirements of Magnesium Deficiency
The first clue to the vitamin-D-sparing effect of magnesium administration was provided by Huffman et al. (1930), who found that a rachitic calf recovered when magnesium carbonate was added to his diet, which was low in calcium but adequate in phosphorus. When it was withdrawn, the abnormal signs recurred. His ribs were soft and broke easily. Calves fed whole milk for 45 days, after which they were given only skim milk and grain [a rachitogenic ration, with a high phosphorus/calcium ratio (Huffman et al., 1930, 1935)], developed hypocalcemia and hyperphosphatemia by 95 days. Magnesium carbonate administration, alone, did not cure the rickets, but when it was given with suboptimal amounts of vitamin D, the biochemical and clinical signs of rickets were corrected. The ash and mineral content of the bones indicated better utilization of calcium and phosphorus when magnesium supplements were given. R. H. Smith (1957, 1961) observed that magnesium supplementation of milk-fed calves that had developed hypocalcemia restored normal serum calcium levels even without vitamin D supplementation and speculated that magnesium might exert its effect on bone/extracellular equilibrium (of calcium). Magnesium-deficient calves had hypocalcemia requiring 70,000 IU/day to correct (R. H. Smith, 1958).
Baby pigs developed rickets on a normally balanced calcium and phosphorus intake (0.8% and 0.6% of the diet, respectively, when they were given less than 100 IU of vitamin D/kg of diet (E. R. Miller et al., 1964b). Most exhibited a moderate fall in plasma magnesium, even on what was shown to be optimal magnesium intakes: 350 ppm of diet (E. R. Miller et al., l965a), particularly in those that developed tetany. All developed hypophosphatemia and hypocalcemia. Balance studies showed that the vitamin-D-deficient pigs absorbed magnesium, calcium, and phosphorus poorly (E. R. Miller et al., 1965b). Doubling the magnesium intake of pigs given no vitamin D improved their weight gain, and prevented the mortality (that resulted in deaths of three of the four vitamin-D-deficient pigs on the standard magnesium intake) but neither prevented their rickets nor corrected their hypocalcemia or hypophosphatemia (E. R. Miller et al., 1964b). Thus, like most human infants, baby pigs require exogenous vitamin D to prevent rickets. Increasing the magnesium intake exerts a partially sparing effect on vitamin D requirements but cannot replace it.
On the other hand, when vitamin D supplements 18-fold higher than the antirachitic amount are given to baby pigs, the greatest strength and elasticity of the femur is obtained with an optimal magnesium intake of 325 ppm (E. R. Miller et al., 1965b). Analysis of rib ash showed no significant effect of low dietary magnesium on percentage of calcium or phosphorus, but a significant reduction in percentage of magnesium. Pigs on low-magnesium intakes, however, exhibited significantly less breaking strength and elasticity (Fig. 12-1). Since the elasticity of the bone is a function of the amount of matrix, the drop in elasticity-but not in bone ash, calcium, and phosphorus of the magnesium-deficient vitamin-D-loaded pigs-reflects the drop in bone magnesium, which is necessary for normal bone matrix formation.
Rats, which are the most commonly employed laboratory animals and with whom most vitamin D and magnesium interrelationships have been studied, differ markedly from the ruminants, pigs, and people as regards their susceptibility to rickets and their response to magnesium deficiency. They do not develop rickets, even when given no vitamin D supplementation, unless they are given diets high in calcium and low in phosphorus (Steenbock diet, cited by Shelling and Asher, 1932). McHargue and Roy (1930) showed that rats fed a normal diet, not supplemented with vitamin D and not exposed to ultraviolet light, remain free of rickets. Au and Raisz (1965) confirmed that rats not supplemented with vitamin D do not develop rickets unless their diets have a high ratio of calcium to phosphorus (0.8% Ca/0.1% P). Rats on high phosphorus to calcium ratios (0.1% P/0.03% Ca) had decreased bone density, but not rickets. The effect of high (14,500 ppm) and normal (6,500 ppm) intakes of calcium fed to groups of rats fed a normal amount of phosphorus (6,100 ppm), but low in magnesium (30 ppm), was studied by Rayssiguier and Larvor (1974a). By the tenth day of the low magnesium intake, all of the rats had hypomagnesemia and low levels of magnesium in their bones. Those that had been given high calcium diets the longest (17, versus 10 days), had the lowest bone magnesium levels. Since high calcium intakes interfere with intestinal absorption of magnesium and increase its urinary output (Reviews: Heaton, 1971; Larvor and Durlach, 1971b; Seelig, 1971), the rats made susceptible to rickets might have been magnesium deficient. Furthermore, vitamin D is necessary for optimal intestinal absorption of magnesium in rats (Meintzer and Steenbock, 1955), as well as in pigs (supra vide) and other species (Schachter and Rosen, 1959; Worker and Migicovsky, 1961) including man. Despite the defective magnesium absorption of vitamin D deficiency, the early rat studies showed high magnesium/calcium ratios in rachitic bones (possibly a reflection of the high osteoid/mineral ratio of such bones). McHargue and Roy (1930), who cited the early studies (Malcolm, 1904; Mellanby, 1926), found that exposure of rats to ultraviolet light for only three to five minutes daily or every other day resulted in better weight gain (than of nonirradiated litter-mates), but in significantly lower bone and total carcass magnesium levels. This work was done before it was realized that magnesium is an essential mineral, and the authors speculated that the beneficial effects of ultraviolet ratio might be the result of eliminating excess magnesium.
Supplementation with vitamin D of rats made rachitic by low phosphorus corrects the hypophosphatemia and heals the rickets (Tanaka and DeLuca, 1974), an effect attributed to a vitamin-D-dependent phosphate transport mechanism (DeLuca, 1976). In 1941, Harrison and Harrison showed that vitamin D increases renal tubular reabsorption of phosphate. Possibly vitamin D's increase of the intestinal absorption of magnesium might also play a role, magnesium deficiency having been shown to exert a phosphaturic effect, even in parathyroidectomized rats (Ginn and Shanbour, 1967). VonEuler and Rydbom (1931) noted the antirachitic effect of adding magnesium to a rachitic diet fed to rats and considered this effect possibly due to magnesium-induced increase in serum phosphatase activity.
In contrast, magnesium deficiency decreases responsiveness to vitamin D in ruminants and rats (R. H. Smith, 1961; Larvor et al., 1964b; Lifshitz et al., 1967a,b). Magnesium-deficient calves required 70,000 IU/day of vitamin D to attain normocalcemia; physiologic doses of vitamin D were not effective (R. H. Smith, 1958). Similarly, Lifshitz et al., (1967b) found that magnesium-deficient rats did not develop a calcemic response to 100 IU of vitamin D a week, but did when the vitamin D dosage was increased 10-fold. Their studies suggested that the poor response of serum calcium in magnesium-deficient rats, to physiologic doses of vitamin D, was due to decreased mobilization of calcium from the skeleton.
Whether impaired mineral mobilization in association with high calcium and vitamin D intake might account for the osteosclerosis seen in rats given intermittent high doses of vitamin D (Storey, 1960), and is mediated by magnesium-deficiency- induced abnormal bone development, deserves study. Storey (1960) observed that large daily doses of vitamin D inhibited endochondrial growth in rats, caused bone resorption and, later uncalcified matrix (osteoid), such as is seen in rickets. When the vitamin D was given intermittently, dense metaphyseal bone was formed in striations, which contained abnormal cartilage, changes resembling those seen in osteopetrosis. It is noteworthy, thus, that comparable changes were seen in infants and children with infantile hypercalcemia, commonly with the supravalvular aortic stenosis syndrome of clinical vitamin D overdosage. Since vitamin D excess causes magnesium loss, it is not surprising that its use (i.e., in milk, which also delivers ample calcium and phosphate) can produce changes in the matrix, such as is seen in magnesium deficiency as well as bone hypermineralization.
Lifshitz et al., (1967b) noted the lag between the time a physiologic dose of vitamin D was given and the calcemic response, and suggested that magnesium's mediating effect might be in its transformation to another form. Since then, it has been demonstrated that vitamin D is hydroxylated to active steroid hormones (e.g., in liver and kidney), and that some of the enzymatic steps require magnesium (Norman, 1968, 1971; DeLuca, 1969; Horsting and DeLuca, 1969; Norman et al., 1975/ 1977). Its deficiency in rats has interfered with the activity of the 1, 25-(OH)2D3 on calcium mobilization from bone, but has not prevented its enhancement of intestinal calcium absorption (Rayssiguier et al., 1974b, 1975).
The cited experimental evidence that magnesium deficiency causes relative refractoriness to vitamin D, very high doses being required for a calcemic response, and that magnesium repletion restores the responsiveness to physiologic doses (supra vide), is reflected by the refractoriness of hypomagnesemic patients to vitamin D. It suggests that the occasional report of correction of vitamin-D-refractory rickets by magnesium might be indicative of the need to evaluate all patients with vitamin-D-unresponsive bone disease for their magnesium status. Conversely, Durlach (1969, 1971) pointed out that patients with latent tetany of magnesium deficiency require less vitamin D when their magnesium deficit is repaired and must have their serum calcium monitored to avoid damage caused by hypercalcemia.
The magnesium/vitamin D/calcium/phosphorus interrelationships are particularly complex. Focusing only on magnesium is unrealistic. Disregarding magnesium is equally unrealistic. Considering only the magnesium/vitamin D interrelationships, if there is a deficiency of magnesium in infancy, for example, there is likely to be impaired response to vitamin D (and to PTH) with resultant hypocalcemia. However, we cannot ignore the hyperphosphatemia of infancy, which is contributed to by the cows' milk and the hypoparathyroidism, and which is enhanced by vitamin D therapy of the hypocalcemia. Thus, in attempting to correct infantile hypocalcemia by calcium loads and calcemic agents a vicious cycle can be established that causes direct loss of magnesium and might damage the area of the renal tubules where magnesium is actively reabsorbed (Fig. 12-4).
12.4.2. Vitamin-D-Refractory Rickets and Osteomalacia
12.4.2.1. Hypophosphatemic Hyperparathyroid Rickets
Hypophosphatemic (vitamin-D-dependent or vitamin-D-refractory) rickets is now the most common cause of rickets in children, and the pathogenesis is still obscure (Cohanim et al., 1972; Brickman et al., 1973). The syndrome was first attributed to hyperparathyroidism secondary to malabsorption of calcium (Albright et al., 1937), and then to an often familial intrinsic renal tubular defect of phosphate reabsorption (B. Robertson et al., 1942; Dent, 1962; Fanconi, 1955; Frame and Smith, 1958; Barbour et al., 1966). Despite the hypophosphatemia, biopsy of the epiphyseal area showed normal content of phosphate (Kuhlman and Stamp, 1964) and also above normal levels of bone alkaline phosphatase, hypertrophic cartilage cells, and thick areas of uncalcified osteoid. Subsequent work has confirmed both hyperparathyroidism (Lafferty et al., 1962; Riggs et al., 1969), usually secondary to intestinal malabsorption (Blackard et al., 1962; Falls et al., 1968; Reitz and Weinstein, 1973), and a genetic X-linked defect in renal tubular reabsorption of phosphate (Glorieux and Scriver, 1972; Glorieux et al., 1973; Scriver, 1973). T. F. Williams (1968) commented on the apparently simple genetics but multiorgan sites of expression in familial hypophosphatemic vitamin-D-resistant rickets. He called for a unifying way to explain the: (1) decreased renal tubular reabsorption of phosphate, (2) decreased intestinal reabsorption of calcium, (3) bony abnormalities, including both osteomalacia and overgrowth, and (4) improvement of calcium absorption and rickets, but not the phosphaturia, with large doses of vitamin D.
Possibly a form of the genetic defect, isolated magnesium malabsorption, is contributory, and might even be a common denominator. This is a point requiring intensive study, and not by measurement of serum magnesium levels. Analysis of bone biopsies for phosphatase and magnesium levels, and metabolic balance studies to ascertain the percentage absorption of orally administered magnesium might be useful. Since such patients are commonly loaded with calcemic agents and phosphates in the attempt to correct their hypocalcemia and hypophosphatemia, and such treatment has increased renal calcinosis, determination of percentage renal retention of magnesium might not be a good index of magnesium depletion. Renal magnesium wasting might result from formation of renal tubular microliths, with damage to the ascending limb of the loop of Henle, where active tubular reabsorption of magnesium takes place. This would perpetuate a magnesium deficit caused by intestinal malabsorption of magnesium.
Magnesium deficiency might be involved in several facets of vitamin D resistance. Both hyperparathyroidism and hypomagnesemia have been implicated in hypophosphatemia (Review: Knochel, 1977), and since familial hypophosphatemia has been found in vitamin-D-resistant rickets in infants and adults (Stickler, 1969; Arnaud et al., 1970; Morgan a al., 1974), there might be a common denominator. There have been several studies that demonstrate abnormal magnesium metabolism and levels, and a few that have shown clinical and biochemical improvement with magnesium therapy.
McCance (1946) reported negative magnesium, calcium, and phosphorus balance in a girl whose vitamin D resistance, osteomalacia, and pseudofractures developed during adolescence. Rosen and Finberg (1972, 1973) found strongly negative magnesium balances in children with active vitamin-D-dependent rickets, which became strongly positive when they had been healed as a result of administration of 25(OH)D3 an active vitamin D metabolite. However, despite negative magnesium balances during the active phase of the disease, serum magnesium levels were within normal limits. Among the conditions found to be associated with low total and ultrafiltrable magnesium levels, reported by Prasad et al. (1961), was a patient with vitamin-D-resistant rickets before treatment.
Administration of magnesium to two children who had rickets, hypocalcemia, and high levels of alkaline phosphatase, despite very high doses of vitamin D2, corrected the biochemical abnormalities and produced X-ray evidence of bone healing (V. Reddy and Sivakumar, 1974). These investigators reported a 5-year-old boy and a 2-year-old girl with rickets, whose hypocalcemia and serum alkaline phosphatase levels of 24.1 and 42.6 Bodansky units failed to respond to several doses of 600,000 IU of vitamin D2. In the boy, serum alkaline phosphatase levels rose further following the high-dosage vitamin D therapy. Severe hypomagnesemia (0.4 mEq/ liter) was then detected and oral magnesium supplementation (10 mEq/day as MgCl2 was started. All biochemical determinants became normal within 4 weeks. The serum magnesium level of the baby girl, who had received 4,000 IU of vitamin D from early infancy, was found to be 0.6 mEq/liter on admission. She was given 600,000 IU of vitamin D daily for 10 days without biochemical improvement. Magnesium supplementation resulted in prompt fall of high levels of serum phosphatase activity, and rises in serum magnesium, calcium, and phosphorus. She was not given the prescribed magnesium at home, and within a month her biochemical abnormalities had recurred. They were promptly corrected on reinstitution of magnesium therapy (Fig. 12-5). Since the diets of these children were not deficient in magnesium, the investigators believe they are probably magnesium malabsorbers.
Rapado et al. (1975) termed the rickets of their 12-year-old patient "magnesium-deficient rickets." She had a long history of polyuria and was found to have nephrocalcinosis with persistent hypercalciuria. After 7 months of treatment with sodium cellulose phosphate (10g/d), her hypercalciuria was corrected, but she developed hypocalcemia, and increased serum alkaline phosphatase. Treatment was then changed to hydrochlorothiazide for two months, after which she developed tetany and osseous pain. Her serum calcium was then 6.9 mg/100 ml, her serum magnesium was 0.5 mEq/liter and her urinary outputs of magnesium and calcium were subnormal. She exhibited subnormal response to PTH. By this time she had signs of overt rickets in wrists and knees. Intramuscular magnesium supplementation (1.5 g/day) for a month resulted in disappearance of the radiologic signs of rickets and correction of the hypomagnesemia and hypocalcemia. On readmission six weeks later, her serum magnesium was again low (1 mEq/liter). On a normal magnesium intake (336 mg she absorbed only 0.2%; thus she represented another instance of magnesium malabsorption. Rapado and Castrillo (1976/ 1980a) have identified another patient with magnesium-dependent rickets, nephrocalcinosis, and who has magnesium malabsorption. Rapado et al. (1975) recommend that patients with vitamin-D-resistant nephrocalcinosis, or failure in response to PTH be evaluated for magnesium deficiency.
Patients with steatorrhea, enteritis, or bypass surgery for obesity have exhibited vitamin-D-refractory osteomalacia (Blackard et al., 1962; Prost et al., 1972; Reitz and Weinstein, 1973; Medalle et al., 1976). Although hypocalcemia is more frequently reported in this disorder, and the hypophosphatemia is commonly attributed to resultant secondary hyperparathyroidism, magnesium deficiency is also common. Reversal of vitamin D resistance has been produced in such patients with magnesium repletion (Medalle et al., 1976).
Although the magnesium status has been shown to influence the response to vitamin D in animals (magnesium deficiency increasing vitamin D requirements) and magnesium is a cofactor in vitamin D conversion to its active steroid-metabolites, its role in vitamin D metabolism in clinical magnesium depletion is not clear. For example, patients who were hypomagnesemic as a result of malabsorption synthesized no less 1,25-(OH)2D3 than did normomagnesemic, hypocalcemic, vitamin D-deficient patients (Lukert, 1976/1980). The active vitamin-D-derived hormones are necessary both for normal intestinal absorption of calcium and for bone calcium turnover (Reviews: Norman, 1974, 1977; DeLuca, 1976). The evidence that vitamin D is necessary for intestinal absorption of magnesium and lowers bone magnesium levels suggests that the active metabolites must also influence magnesium metabolism, and abnormality in vitamin D metabolism probably influences bone integrity as a function, not only of changes in handling of calcium but of magnesium. Avioli et al. (1967) found increased levels of vitamin D metabolites without calcium absorptive activity in hypophosphatemic rickets. How such metabolites influence magnesium absorption or bone levels has not been reported.
Vitamin D increases renal tubular reabsorption of phosphorus (Harrison and Harrison, 1941). A vitamin-D-dependent intestinal phosphate-transport mechanism (DeLuca, 1976) can partially explain the hypophosphatemia of vitamin-D-refractory rickets. Association of abnormal vitamin D metabolism with secondary hyperparathyroidism (Arnaud et al., 1970) supports the premise that hyperparathyroidism, whether secondary to intestinal malabsorption or to abnormal vitamin D metabolism, contributes to impaired phosphate reabsorption by the kidneys. Clinical evidence of the importance of the abnormality of vitamin D metabolism in hypophosphatemic rickets has been obtained by demonstration of response of patients with this disorder to the 25-(OH)D3 (Rosen and Finberg, 1972, 1973), to the 1,25-(OH)2D3 (Fraser et al., 1973; Avioli and Haddad, 1973; Balsan et al., 1975) and to 1,a(OH)D3 (Balsan et al., 1975; Rosen and Finberg, 1975/1977). The active metabolites, like the parent substance, vitamin D3, correct the malabsorption of calcium and the impaired bone mineralization, but do not influence the form of the disease that is characterized by intrinsic defective renal tubular phosphate reabsorption (Brickman et al., 1973; Glorieux et al., 1973). Possibly relevant to this partial failure of treatment is the demonstration of phosphaturia in magnesium-deficient rats (despite their hypercalcemia, and even in those that were parathyroidectomized) by Ginn and Shanbour (1967).
12.4.2.2. Hyperphosphatemic, Hypoparathyroid Osteopenia
A paradoxical facet to vitamin D resistance is that, in addition to its association with hyperparathyroidism and hypophosphatemia, magnesium-reversible vitamin D refractoriness has long been recognized in hypoparathyroidism with hyperphosphatemia (Homer, 1961; K. Jones and Fourman, 1966; Harrison et al., 1967). A child with idiopathic hypoparathyroidism who had hypocalcemia and low serum alkaline phosphatase levels responded to PTH with phosphaturia, but without correction of her hypocalcemia (Rösler and Rabinowitz, 1973). She was then given up to 600,000 IU of vitamin D and 3-4 g of calcium lactate for 13 weeks, and then dihydrotachysterol, without raising her blood calcium level. Her serum magnesium was then found to be 0.5 mEq/liter, and she developed convulsions and tetany. Magnesium repletion produced rapid improvement. Medalle and Waterhouse (1973) reported a biochemical picture of hypoparathyroidism, hypocalcemia and hyperphosphatemia in a patient with severe magnesium depletion of chronic alcoholism, and suggested that magnesium deficiency be considered in the differential diagnosis of hypoparathyroidism, pseudohypoparathyroidism, and renal failure. Their patient did not exhibit a normal phosphaturic response to PTH while she was magnesium depleted. Magnesium therapy corrected her hyperphosphatemia promptly; correction of her hypocalcemia was more gradual. This finding suggested to the investigators that hyperphosphatemia does not occur unless the magnesium depletion is severe. They noted that it occurs less frequently than does hypocalcemia in magnesium-deficient patients, and has been reported with selective experimental magnesium deficiency (Shils, 1969a) or with isolated magnesium malabsorption (Dooling and Stem, 1967; Skyberg et al., 1967; 1968; Stromme et al., 1969; Nordio et al., 1971).
It might be well, also, to ascertain whether the high vitamin D requirements of low-birth-weight infants (who are also subject to hypoparathyroidism and hyperphosphatemia) might be contributed to by magnesium inadequacy. Vitamin-D-resistant rickets of biliary atresia, which is responsive to 25-OHD3 (Daum et al., 1976; Rosen and Finberg, 1975/1977), might also be responsive to magnesium, magnesium deficiency having been demonstrated in such infants (A. Kobayashi et al., 1967, 1974). On the other hand, a defect in intestinal absorption of magnesium, as well as of calcium, has been found in idiopathic hypoparathyroidism, which improved following treatment with 25-OHD3 (Rosen and Finberg, 1975/1977).
12.4.3. Other Abnormal Function of, or Response to, Parathyroids
Magnesium deficiency affects parathyroid hormone (PTH) secretion, and target organ response, and several syndromes are associated primarily with skeletal abnormalities that might have abnormal magnesium metabolism as a common denominator. For example, idiopathic hypoparathyroidism and pseudohypoparathyroidism and variations of these disorders that have been given cumbersome names (e.g., pseudopseudohypoparathyroidism, pseudohypohyperparathyroidism) might be explicable on the basis of different phases and degrees of magnesium deficiency and its influence on response to calcemic or hypocalcemic therapy. Bronsky (1970) criticized the nomenclature used for the variations in disorders that are associated with Albright's osteodystrophy (brachydactyly, stocky body, and round face) or dyschondroplasia with soft tissue calcinosis. Selected from his tabulated forms of parathyroid disease are several that might derive from abnormalities of magnesium metabolism, or from maternal, perinatal, or later dietary imbalances (Table 12-1). Patients with osseous or soft tissue calcinosis frequently have close relatives with parathyroid disease or convulsive disorders, which is suggestive of a possible contributory magnesium deficit. Bronsky (1970) stressed the relationship of steatorrhea to parathyroid disease (both hypersecretion and resistant hypoparathyroidism), an observation that supports the concept of underlying magnesium deficiency. Discussed earlier are gestational hyperparathyroidism and neonatal hypoparathyroidism and their likely interrelationships with magnesium deficiency.
Albright et al. (1942), who first described the pseudohypoparathyroid dyschondroplasia syndrome, noted that patients with this disorder had convulsions themselves and had siblings with epilepsy, were mentally retarded, and were resistant to high doses of calcemic agents. These manifestations, and the histories of infantile respiratory distress (reported in the original and subsequent cases) resemble those of infants who developed cardiovascular and/or renal abnormalities that are speculated also to be related to magnesium deficiency. The patient reported by C. Lowe et al. (1950), who had an early symptom-complex very much like infants with hyperreactivity to vitamin D, early developed signs of vitamin-D-resistant rickets and spontaneous fractures, such as are seen in hypophosphatasia (related to magnesium deficiency). When she was found to have steatorrhea, the authors speculated that malabsorption might have contributed to her disorder; the malabsorption certainly could have caused magnesium depletion. Talbot et al. (1954) described the syndrome in twins, and commented that hypocalcemic symptoms of this disorder usually date from the neonatal period, further suggestive evidence of magnesium deficiency.
An infant who died at two months of age, who had neonatal hypoparathyroid, hypocalcemia, and hypomagnesemia, had had similar signs, persistent diarrhea, and renal tubular acidosis, but no X-ray evidence of rickets or soft tissue calcinosis. PTH injection evoked a phosphaturic and calcemic response, but lowered her serum magnesium to subnormal levels. Calcium therapy further lowered it, and caused hypercalcemia (Taitz et al., 1966). At autopsy, neither parathyroid nor thymic tissue was found, and there were intraluminal renal tubular calcium deposits. The bones were not examined. Whether this case reflects profound prenatal parathyroid suppression, or a genetic defect, complicated by a metabolic disorder as proposed by the investigators, and whether longer survival would have resulted in overt skeletal abnormalities, is not possible to aver.
The manifestations of the disorders categorized as "idiopathic" hypoparathyroidism and pseudohypoparathyroidism or dyschondroplasia, predominantly on the basis of presence or absence of phosphaturic response to PTH, were compiled by Bronsky et al. (1958). Their tabulation of symptoms and signs of patients diagnosed as "idiopathic" or "pseudohypoparathyroid" (Table 12-2) and their comment that both are idiopathic, the cause not being known, is valuable. It should be noted, however, that current emphasis is on failure of both kidneys and bone of pseudohypoparathyroid patients to respond to PTH, and on failure of 3',5' cyclic adenine monophosphate (cAMP) response (Chase et al., 1967, 1969; Drezner et al., 1973). That this abnormality might underlie the refractoriness of bones and kidneys to PTH (Coburn et al., 1972) points toward a possible underlying magnesium deficiency, the synthesis of cAMP having an absolute magnesium requirement (Sutherland et al., 1968).
Evidence has been presented that prenatal magnesium deficiency can contribute to neonatal hypoparathyroidism, and that subsequent dietary imbalances might predispose to permanent damage. Not only the cardiovascular system, but the skeletal and renal systems can be affected, the manifestations depending on combinations of genetic metabolic abnormalities, dietary imbalances, and treatment. Perhaps the higher incidence of thickened calvaria and the much higher incidence of soft tissue calcinosis and mental retardation of the pseudohypoparathyroid group than of the "idiopathic" group might reflect a (postulated) greater magnesium deficit and a consequent need for higher doses of calcemic agents to correct hypocalcemia, with resultant signs resembling those of infantile hypercalcemia. The coarse trabeculation of the short bones of this group of patients, in fact, resembles that seen in experimental magnesium deficiency. Also, the occasional exostosis or tumorlike growth on long bones resembles that seen in patients with hypophosphatasia or osteogenesis imperfecta (speculated to be contributed to by magnesium deficiency) and seen in experimental magnesium deficiency. Slipped epiphyses, such as have been reported in a few patients with magnesium deficiency and that are in accord with the epiphyseal abnormalities of experimental magnesium deficiency have also been reported in different forms of pseudohypoparathyroidism (Bronsky et al., 1958; Frame et al., 1972). Young patients with the disorder, termed "pseudohypohyperparathyroidism," who had osteosclerosis in the skull, osteitis fibrosa in long and flat bones, and slipped epiphyses, have had serum magnesium levels reported within normal limits (Frame et al., 1972). The adolescent girl, who had had resistant rickets and subsequent nephrocalcinosis from infancy, and who developed osteitis fibrosa cystica and parathyroid adenomas after years of high-dosage calcemic therapy, had both hypercalcemia and hypermagnesemia on admission to the hospital (W. Thomas and Fry, 1970). The past history of nephrocalcinosis should predispose to renal magnesium wastage, which either did not exist in this patient or was masked by PTH-mobilization of bone minerals preoperatively. More intensive studies of the magnesium status of such patients are necessary to clarify whether cellular magnesium deficit might exist, despite lack of hypomagnesemia. Studies of magnesium metabolism in members of families with either hypo- or hyperparathyroidism that has a genetic component (whether the complete syndrome exists, or only some of the manifestations) might be fruitful.
The condition termed "renal rickets" has long been known to be associated with severe skeletal distortions, acidosis, renal calcinosis, hyperparathyroidism, and mental and growth retardation (Shelling and Remsen, 1935; Price and Davie, 1937). The child reported by Price and Davie (1937) is of particular interest in building up a case for primary magnesium deficiency, since he was the product of the seventh pregnancy, and had been born the year after a miscarriage and the year before two additional miscarriages. As has been discussed, frequent pregnancies are likely to predispose to fetal magnesium deficiency and to spontaneous abortions. This child had generalized osteoporosis and alternate sclerosis and rarefaction of the skull at the age of 14, florid rachitic changes at the extremities, slipped epiphyses, renal damage, deafness, and evidence of mental retardation. At autopsy, it was found that the radiologic diagnosis of slipped femoral epiphysis was incorrect; he actually had collapse of the metaphysis of the neck of the femur, and bone was replaced by a mixture of fibrous tissue and cartilage. All four of his parathyroids were hyperplastic. There were numerous small foci of calcification in his kidneys. These investigators question whether it is necessary to be certain that the renal lesion has preceded the other findings for a diagnosis of renal rickets to be made, as was the case in the similar boy reported by Shelling and Remsen (1935). That boy had hypercholesterolemia, hypertension and arteriosclerosis, and hyperphosphatemia despite parathyroid hyperplasia, elevated PTH levels, and skeletorenal lesions much like those of the child reported by Price and Davie (1937).
In 1927, L. Parsons described five children with fragile bones and rickets secondary to celiac disease. He noted that the skeletal deformities usually do not develop until the age of seven years. One of his patients had blue sclerae similar to that seen in osteogenesis imperfecta, which gradually became normal in color as the malabsorption improved. Spontaneous pseudofractures were sometimes seen, severe osteoporosis, and persistently fragile bones, even after control of the malabsorption, and despite treatment with cod liver oil. These manifestations are of interest because of their similarity to those of experimental magnesium deficiency and to diseases speculated to be contributed to by magnesium depletion. Prost et al. (1972) have described osteomalacia secondary to malabsorption in adults. They correlated osteomalacia and pseudofractures with hypomagnesemia in two instances, and recommended evaluation of the magnesium status with a view to its repair, in an effort to restore vitamin D responsiveness in such patients.
12.4.4. Osteopetrosis or Osteosclerosis and Hyperreactivity to Vitamin D
12.4.4.1. High Vitamin D and Calcium/Low Magnesium
Skeletal changes similar to those seen in magnesium-deficient animals given diets relatively high in calcium and vitamin D, or in hypervitaminosis D studies, are seen in clinical osteopetrosis. Storey (1960) has reviewed the X-ray, histological, and biochemical findings of this disease. Pathognomonic is alternation of radiopaque and radiolucent transverse bands running parallel to the epiphyseal cartilage of the long bones, and to the surface of other bones. Histological studies have shown that the bone is increased in amount, but abnormal. There is some normal bone, islands of densely calcified cartilage near the epiphyses, and areas of osteoid tissue so poorly calcified as to resemble rickets. Microradiographs show areas of high and low bone density, and exaggerated thick radiopaque "cementing" lines on the surface and concentrically around immature Haversian systems. Intense bone resorption is also occasionally seen. These changes are often accompanied by generalized calcinosis of arteries, kidneys, ligaments and tendons, and other soft tissues. Biochemical changes are inconstant, depending on the stage of the disease. Serum calcium levels are usually normal, but hypercalcemia has been reported. Serum phosphorus is often low, with a Ca X P product suggestive of vitamin-D deficient rickets, or sometimes of vitamin-D-refractory rickets. Storey (1960) confirmed the bone changes of "hypervitaminosis D rickets" in rats, as described by Ham and Lewis (1934), who found flattened, thinned epiphyses, numerous thickened trabeculae, and matrix ranging from normal to poorly calcified, and then possibly "compensatory" excess osteoid. When he gave high doses of vitamin D intermittently to rats (Storey, 1960), the bone changes were very much like those seen in clinical osteopetrosis. The base of the skull became extremely dense and thick. Storey (1960), puzzled over the bone changes caused by excess vitamin D in his own and other studies and considered mediation by hormonal responses, calcium, phosphorus, and "as yet unelucidated systemic disturbances." The similarity of the excess vitamin-D-induced changes of the epiphyses, trabeculae, and matrix, to those described by Bernick and Hungerford (1965) in magnesium-deficient rats suggests that magnesium loss from bone, caused by excess vitamin D, might be a contributory factor.
Magnesium balance data were obtained in a series of balance studies (done over an eight-month period) in an infant with roentgenologic evidence of osteopetrosis but with biochemical evidence of hypophosphatemic rickets: marginal hypocalcemia, hypophosphatemia, and elevated serum alkaline phosphatase (Pincus et al., 1947). Although the authors did not comment on the magnesium findings, analysis of their data shows that in the preliminary test period (at two and one-half months of age) the baby retained 10 times as much calcium as magnesium, and her calcium/magnesium absorptive ratio was 7/1. Two months after her vitamin D2 supplementation was increased severalfold over the usual dose, her retention of calcium was 16 times that of magnesium, and she absorbed 10 times as much calcium from the gut. During the last balance period (at 10 months of age) when her magnesium intake had been increased to 553 mg per day and her calcium intake had also been increased but proportionally less, the ratio of intestinal calcium to magnesium was 6/1. Her retention of Ca/Mg, however, was 9/1, a greater percentage of the absorbed magnesium being excreted in the urine. Her serum alkaline phosphatase had fallen by that time to hypophosphatasia-levels, her serum calcium remained marginally low, but her serum phosphorus had risen to 5.5 mg/100 ml. A trial of parathyroid extract transiently increased the serum calcium to within normal limits, and the serum phosphorus gradually fell to 3 mg. After 6 weeks, the child became refractory to parathyroid treatment. She died at 16 months, and her osteopetrosis was confirmed at autopsy
Infantile hypercalcemia is associated with osteosclerosis. The first such patient reported was a dwarfed infant with hypercalcemia, cardiovascular and renal calcinosis, and mental retardation (Lightwood, 1932). What was then a rare syndrome appeared much more commonly in the literature in the 1950s, during an era of excessive fortification of milk with vitamin D in the British Isles (Review: Seelig, 1969). The skeletal abnormalities were less commonly reported than was the severe cardiovascular, renal, and mental damage, which was termed the supravalvular aortic stenosis syndrome (SASS, Editorial, Br Med J, 1956). Fanconi and Girardet (1952) described an infant with the full syndrome. British babies were then reported with radiographic evidence of excessive deposition of sclerotic bone at the base of the skull, in periorbital bones, at ends of long bones, and at the borders of the vertebrae (Creery, 1953; Russell et al., 1954; Dawson et al., 1954; Lowe et al., 1954; Stapleton and Evans, 1955; Schlesinger et al., 1956; Joseph and Parrott, 1958). The amount of vitamin D estimated to be consumed by the affected children ranged from 1000 to 3200 IU, an amount that is not infrequently provided by the American diet. And, in fact, these lesions have not been limited to the British babies. The syndrome has been described in continental Europe and in America, the cardiovascular anomalies more frequently, the skeletal changes less frequently. Shiers et al. (1957) reported four children from one-and-a-half to almost five years of age, all of whom had roentgenologic evidence of osteosclerosis and other signs of hypervitaminosis D, but none of whom had histories of its excessive consumption. One had multiple bands of sclerosis parallel to the growing ends of the long bones, and distorted shafts; one had increased skull density, particularly at the base, with increased density of vertebral and carpal bones and of epiphyses, and one had rachitic-like lesions of the ends of the long bones but generalized osteosclerosis. The authors noted that the most heavily sclerosed bone had been laid down in utero. The oldest child, who was also hypothyroid, had very heavy osteosclerosis, particularly in the cranial and facial bones. All bones were affected, with bands of varying density. Three infants, who had been born prematurely, developed the classic signs of severe hypercalcemia by 6 months of age, and were found to have osteosclerosis at 10 to 17 months of age (Singleton, 1957; Daeschner and Daeschner, 1957; Snyder, 1958). None had been given more than 1000 IU of vitamin D as supplements (in addition to that provided by milk and other fortified foods). A Swiss child of low birth weight was born to a mother who later developed diabetes mellitus (a condition associated with low magnesium levels) and developed the full-blown syndrome by 5½ months of age after high-dosage vitamin D (Illig and Prader, 1959) Another infant who was small at birth, born to a mother who had taken 1000 IU vitamin D daily during much of her pregnancy, developed the syndrome at 4 months of age (Fraser et al., 1966). Others, who developed the classic signs of hypercalcemia, SASS, and osteosclerosis at 9 to 18 months of age, were normal-sized at birth and had not been given high-dosage vitamin D supplements (O'Brien et al., 1960; N. David et al., 1962; Garcia et al., 1964; D. Fraser et al., 1966). The youngest infant with hypercalcemia and osteosclerosis had not had high-dosage vitamin D but had been given supplemental calcium (Wilkerson, 1964). Hyperreactivity to vitamin D is suspected in these children.
Infants and children have developed the complete hypercalcemic syndrome, including osteosclerosis, after massive intermittent doses of vitamin D (Amann, 1959; Manios and Antener, 1966). A child who had received excessive daily vitamin D supplements from his third through fifth years of age developed periarticular calcification and hypertension as well as osteosclerosis. He died, two years after his excessive supplements had been stopped, with renal failure and coronary atherosclerosis (DeWind, 1961).
Search for possible prenatal factors in the pathogenesis of infantile hypercalcemia and the SASS, led to studies of pregnant rabbits overdosed with vitamin D. W. Friedman and Mills (1969) found that some of the young had premature closure of the cranial bones, osteosclerosis, and palatal abnormalities similar to those seen in infants and children with infantile hypercalcemia and the SASS. Rowe and Cooke, (1969), considering the role of maternal vitamin D in the genesis of the excessive fetal mineralization in the rabbits (W. Friedman and Mills, 1969; W. Friedman, 1968), commented that mothers of children with the SASS had not usually had histories of vitamin D overdosage during pregnancy. They noted that Friedman and Mills (1969) had considered the possibility of acquired decreased tolerance of vitamin D. They suggested that an infant who had undergone excessive mineralization in utero might be unduly susceptible to both hypercalcemia and osteopetrosis thereafter. It should be noted, here, that early studies of the effects of supplementing pregnant women with only moderate doses of vitamin D showed that the fetuses tended to have narrower cranial sutures and greater bone density than did the fetuses of control nonvitamin-D-supplemented mothers (Finola et al., 1937; Brehm, 1937; Review: Seelig, 1978). Rowe and Cooke (1969) proposed that there might be a failure of regulating mechanisms for blood calcium in infants with SASS and osteosclerosis, and that there is probably a multifactorial basis for the difference in susceptibility to the disease. A factor that should be considered is the possible role of magnesium deficiency: gestational, magnesium malabsorption, or vitamin D induced. The ranges of susceptibility to vitamin D toxicity (Fanconi, 1956), and the magnesium loss caused by excess vitamin D should also be taken into account.
Intermittent magnesium treatment of the constipation characteristic of infantile hypercalcemia has been mentioned by some of the investigators of infantile hypercalcemia (Creery, 1953; Lowe et al., 1954; Forfar, thesis). Stapleton and Evans (1955) noted that a hypercalcemic infant fed a formula free of calcium and magnesium exhibited a steady drop in serum magnesium levels (to 1.4 mEq/liter). Lowe et al. (1954) reported hypomagnesemia in a mild case and hypermagnesemia in a severe case. Metabolic balance studies of severely hypercalcemic infants showed that they were in magnesium equilibrium (McDonald and Stapleton, 1955), only slightly positive (+ 1.3 mg/kg/day) or negative (Forfar, thesis). Fellers and Schwartz (1958), who studied two infants with severe hypercalcemia even when all vitamin D was removed from the diet, and who suggested that the disease is caused by abnormal vitamin D metabolism (Fellers and Schwartz, 1958b), reported that when calcium and vitamin D were deleted from the diet, the children went into strongly positive magnesium balance. These data suggest that magnesium deficiency may be part of this syndrome since infants should be in strongly positive magnesium balance (Seelig, 1964, 1971). Dalderup (1960) was the first to propose that magnesium deficiency might be contributory to this disorder.
The cited metabolic balance study by Pincus et al. (1947) supports the premise that magnesium malabsorption might be an initiating disorder that might contribute to hypophosphatemic rickets. Vitamin D, given to infants whose bone matrix is abnormal because of magnesium deficiency, might lead to hypermineralization, such as is produced in rats on high-dosage vitamin D plus calcium. The development of hypophosphatasia after 8 months of high-dosage vitamin D in the infant studied by Pincus et al. (1947), and the hypophosphatasia found in infantile hypercalcemia with hyperreactivity to vitamin D, suggest that intensification of magnesium deficiency by excessive vitamin D might be at fault, alkaline hypophosphatasia also being characteristic of magnesium deficiency.
However, once hypercalcemia is part of the clinical syndrome, it should be corrected before attempting to correct the magnesium deficiency with a parenteral magnesium load. Alkaline and pyrophosphatases (which destroy the calcification-inhibiting polyphosphates and pyrophosphates) are found, not only in bone but in the kidneys, cardiovascular, and other soft tissues. Since the phosphatases are magnesium dependent, administration of magnesium (in the face of hypercalcemia) might increase the risk of metastatic calcification, as had been suspected by the physicians who treated hypercalcemic infants (supra vide). Whittier and Freeman (1971) have provided experimental evidence that administration of magnesium to rats made hypercalcemic by hypervitaminosis D did in fact increase renal and myocardial calcification.
Congenital osteopetrosis need not be associated, however, with hypercalcemia. Rosen and Haymovits (1972) have reviewed the evidence that the disease is characterized by impaired bone resorption, and have speculated that a defect in lysosomal functions might be a significant factor in its pathogenesis. They demonstrated increased levels of the hepatic lysosomal enzyme, β-glycerophosphatase (the significance of which is unclear), and increased frequency of hepatic electron-dense mitochondrial particles. Whether these granules are comparable to those reported in myocardial mitochondria in magnesium deficiency and whether they are an indication of magnesium deficiency is speculative.
12.4.4.2. Magnesium/Calcitonin Interrelationships in Osteoporosis
In considering the effect of vitamin D and calcium supplementation to pregnant women, the active transport of calcium across the placental barrier and the effect of high calcium levels on calcitonin (CT) secretion should also be taken into account. Acute hypercalcemia (in rats) has lowered the CT content of thyroid C cells (Gittes et al.., 1968), and has increased plasma immunoreactive CT levels in several species of animals (Littledike et al., 1972). There is direct evidence that the hypercalcemia caused by excessive vitamin D (in cows) increases CT release (Young and Capen, 1970). In the gray lethal mouse, which develops osteopetrosis, it has been proposed that the primary lesion is hyperplasia of thyroid C cells, with overproduction of CT (D. Walker, 1965, 1966). There is evidence that CT not only inhibits bone resorption (Johnston and Deiss, 1966; Bélanger and Rasmussen, 1968; Raisz et at., 1968; Baylink et al., 1969; Hirsch and Munson, 1969), but that it also increases bone calcification, growth, and repair (Wase et al., 1967; Pallasch, 1968; Ziegler and Delling, 1969; Delling et al., 1970; Gaillard and Thesingh, 1968; Matthews et al., 1972; Salomon et al., 1973). Fetuses infused with calcium secrete CT (Littledike et al., 1972; Garel et al., 1973, 1974, 1976; Garel and Barlet, 1974) and the high fetal and cord CT levels are presumed to play an important role in normal bone growth and calcification (Samaan et al., 1973, 1975). Thus, it seems likely that hypercalcemia of fetuses of mothers given excessive vitamin D might cause abnormally high fetal CT levels and increase bone mineralization. It is possible that low fetal magnesium levels, such as is postulated to be not uncommon, also increases CT secretion. The influence of the fetal magnesium/calcium ratios on the PTH/CT responses will influence the nature of the changes induced in fetal and infantile bone.
12.5. Other Genetic Bone Diseases and Possible Role of Magnesium
12.5.1. Osteogenesis Imperfecta
The similarity of the bone lesions in young of rats given excessive vitamin D during pregnancy to those of osteogenesis imperfecta, the magnesium depletion caused by hypervitaminosis D, and the infantile osteopenia and spontaneous fractures seen in infants likely to have magnesium deficiency (supra vide), suggest that the magnesium status of members of families with osteogenesis imperfecta be explored. It is conceivable that familial malabsorption or renal wastage of magnesium might be contributory to the familial occurrence of osteogenesis imperfecta.
Whether osteogenesis imperfecta is a separate entity from the severe early form of hypophosphatasia, the bone lesions of which are indistinguishable from it, is not yet certain. The essential difference is in the serum phosphatase levels that have been reported. Hansen (1934) confirmed earlier reports that patients with osteogenesis imperfecta do not have the low serum alkaline phosphatase levels that are characteristic of hypophosphatasia. However, he analyzed tissues of a child who died of the disease, without having received unusual medication, and found almost complete absence of phosphatase in the periosteum and subperiosteal structures, where it is normally abundant. Solomons and Styner (1969) studied 28 patients (2 days to 14 years of life) with this disease and found the collagen biopsies completely prevented mineralization at pH 7.4, and that pyrophosphatase in the presence of magnesium (3 X10-3) markedly reduced the inhibition. Addition of magnesium without the enzyme partially reduced the inhibition of mineralization. They reported that bone from patients with osteogenesis imperfecta had much higher levels of pyrophosphate than did normal bone. This excessive pyrophosphate could be almost completely removed by in vitro treatment with pyrophosphatase plus magnesium. They also reported significantly higher than normal serum pyrophosphate levels in serum and urine, a finding not corroborated by R. Russell et al. (1971), who found higher than normal plasma levels only in hypophosphatasia. (The latter investigators, however, cautioned that plasma pyrophosphate levels might not be in equilibrium with that in bone or other tissues.) In view of the difference in pyrophosphate levels reported by the two groups, it is not possible to evaluate the significance of Solomons' and Styner's (1969) clinical report that administration of magnesium salts (2-6 mg/kg) to four patients with osteogenesis imperfecta lowered their serum and urine pyrophosphate levels toward the normal range.
J. Albright and Grunt (1971) studied magnesium balance (among other elements) in five children with osteogenesis imperfecta before and after fluoride treat ment. All had negative magnesium balances, which were not affected by the fluoride. Riley and Jowsey (1973) treated three patients with magnesium oxide (15 mg/ kg/24 hr) with only minor changes in bone formation and resorption, as measured by microradiography of iliac crest biopsies (Table 12-3). Whether the slight increases in bone formation and increases in bone formation and resorption noted in the two children with the severe form of the disease might have continued with prolongation of magnesium therapy would have been of interest. The older child, whose disease was less severe, and whose microradiographic studies showed less abnormal bone turnover, showed a drop in bone resorption (but still to within normal limits) but also a decrease in new bone formation. Intestinal and renal handling of magnesium should be correlated with bone response.
Benign hyperplastic callus formation, which simulates osteosarcoma, has been reported in patients with osteogenesis imperfecta. Banta et al. (1971) reviewed 21 published cases, and 2 of their own, of such superabundant callus (usually of the tibia or femur but sometimes of the pelvis) that led to amputation for sarcoma in several instances. Replacement of muscle tissue by the extensive fracture callus was consistent with myositis ossificans. One of their patients (a young man of 22) also had bilateral dislocation of the radial heads and ankylosis of the spine. These abnormalities are noted because of the demonstration of exuberant growth of the femur, simulating osteosarcoma, of magnesium-deficient rats, and of the possibility that slipped epiphyses and chondrocalcinosis, including spondylitis, might be related to magnesium deficiency. Further evidence of abnormalities in collagen of patients with osteogenesis imperfecta derives from studies of skin collagen (Haebara et al., 1969; C. Stevenson et al., 1970; R. Smith et al., 1975) and bone collagen and matrix proteins (Haebara et al., 1969; Dickson et al., 1975). Thin scleral collagen has been suggested as a factor in the characteristic blue sclerae. If the abnormality in bone matrix is similar to that produced by experimental magnesium deficiency (Bernick and Hungerford, 1965; Trowbridge and Seltzer, 1967), and if the propositus and his close relatives can be shown to absorb or retain magnesium abnormally, we might have another clue to the pathogenesis of this disease.
Another fragment of evidence that magnesium deficiency might be participatory is the aminoaciduria detected in some patients with osteogenesis imperfecta and in members of their families (Chowers et al., 1962; Brigham and Tourtelotte, 1962; Summer and Patton, 1968). Five children with osteogenesis imperfecta were born to three families, almost all the members of which had aminoaciduria (Chowers et al., 1962). The authors had investigated the amino acid excretory patterns of the families because of the frequent association between bone-wasting diseases and renal tubular dysfunction (e.g., osteomalacia, rickets, Fanconi syndrome, and hyperparathyroidism). Aminoaciduria has been produced in animals by experimental magnesium deficiency and is seen in patients with hyperreactivity to vitamin D (Fanconi and Girardet, 1952) or with intestinal malabsorption (Muldowney et al., 1968), both conditions in which magnesium deficiency is demonstrable or suspected. Abnormal amino acid urinary output has been repeatedly demonstrated (Seelig and Berger, unpublished observation) in a woman with rapidly progressive osteoporosis, latent tetany of magnesium deficiency (Seelig et al., 1975) and renal magnesium wastage (Seelig et al., 1976/1980). The amino acid urinary excretory pattern of infants who have been given excessive vitamin D or who have hyperreactivity to vitamin D has rarely been reported. Drummond et al. (1964), however, ascertained that infants with familial hypercalcemia and nephrocalcinosis have abnormal tryptophan metabolism, termed the "blue diaper syndrome." This abnormality is of interest, since comparable abnormal metabolites of tryptophan are excreted in vitamin B6 deficiency or abnormality, and pyridoxine enzymes are magnesium dependent (Review: Durlach, 1969b)
Osteogenesis imperfecta, like hypophosphatasia, abnormalities in vitamin D or magnesium metabolism, and congenital heart diseases that have been correlated with either or both of these metabolic abnormalities, can be isolated or familial. It is of interest that the bone and cardiac disorders have been seen in the same patient, sometimes in association with renal calcinosis. For example, Coleman (1959) reported a baby with osteogenesis imperfecta, who died with nephrocalcinosis and thrombosis, among a series of 24 with infantile hypercalcemia, whose ECG changes (ST-T abnormalities) were not related to serum calcium levels. Examination of the ECG data shows similarities to those reported in conditions associated with magnesium deficiency (Review: Seelig, 1969a). It has been suggested that idiopathic hypertrophic subaortic stenosis might similarly be associated with hypercalcemia (McFarland et al., 1978). Whether the growth retardation and skeletal abnormalities (particularly of the face and base of skull, leading to cardiofacies, and of the chest) that have been seen in cardiac outflow abnormalities (Chapter 4) are similarly mediated cannot be averred. Investigation of the metabolism of magnesium and of vitamin D of the propositus, and especially of infant siblings and mother, might provide insight into the etiology of these forms of combined cardiac and skeletal abnormalities.
It should be recalled that infantile hypercalcemia is frequently associated with the supravalvular aortic stenosis syndrome and with other cardiac outflow abnormalities. It is thus provocative that osteogenesis imperfecta has been reported in patients with aortic coarctation (Remigio and Grinvalsky, 1970) and in patients with valvular abnormalities requiring correction by open heart surgery (Criscitiello et al., 1965; Heppner et al., 1973; Wood et al., 1973; Waters et al., 1977). Perhaps most directly suggestive of the role of gestational magnesium deficiency in the pathogenesis of the combined congenital abnormalities of osteogenesis imperfecta, valvular disease, and aortic coarctation, are the two infants born with these disorders to a young woman who had had multiple pregnancies at short intervals (Remigio and Grinvalsky, 1970). They were the products of her ninth and tenth pregnancies, the seventh and eighth having terminated as spontaneous abortions. Such frequent pregnancies have been shown to be associated with maternal magnesium depletion. However, there might well have been a genetic predisposition to skeletal abnormalities, since the first two siblings had abnormalities of their hips. McKusick (1966) and Shoenfeld et al. (1975) have cited premature arteriosclerosis in osteogenesis imperfecta, another hint at possible underlying magnesium deficiency that is probably caused by defective ability to absorb or retain magnesium.
Hyperparathyroidism has also been associated with magnesium loss, and thus the coexistence of hyperparathyroidism and osteogenesis imperfecta tarda in women in their late forties or early fifties (Goldzieher et al., 1957; Quay et al., 1968; Salti et al., 1973; Woolfson et al., 1975) provides still another piece of circumstantial evidence linking magnesium deficiency with this form of osteopenia. Whether decreased estrogen secretion, which antagonizes parathyroid hormone activity, allows for an occult disorder to become overt in patients with mild forms of this disease is speculative.
Patients with osteopenias are commonly treated with high-dosage calcemic agents, which increase both magnesium loss and extraskeletal calcification. Thus, the combination of bone defects with damage to such organs as the heart, arteries, and kidneys, and ectopic calcification is explicable on the basis of a primary magnesium deficiency that increases susceptibility to toxicity of calcemic agents and ectopic calcification.
12.5.2 Hypophosphatasia
The term "hypophosphatasia" has been applied to the inborn error of metabolism that is characterized by defective bone mineralization, associated with low serum alkaline phosphatase activity, and high urinary output of phosphoethanol amine (Reviews: Fraser, 1957; Currarino, 1957). Most of the reports of this condition also indicate hypercalcemic values. No data have been found on magnesium levels, but there is reason to suspect that magnesium deficiency might be contributory to development of this syndrome.
Experimental magnesium deficiency causes low levels of alkaline phosphatase activity in bone, as well as in serum. Rats surviving few to 28 days of magnesium deficiency, and then repleted, had fragile bones thereafter (Duckworth et al., 1940), such as are seen in adults whose hypophosphatasia is diagnosed late (Fraser, 1957). Low levels of bone alkaline phosphatase have been reported in patients with hypophosphatasia (Rathbun, 1948; Sobel et at., 1953; Engfeldt and Zetterstrom, 1954; Schlesinger et al., 1955; Currarino et al., 1957). Without optimal amounts of alkaline phosphatase in bone, its mineralization is inhibited, since alkaline phosphatase is necessary for local destruction of mineralization inhibitors, such as polyphosphates and pyrophosphates. Additionally, high levels of phosphates intensify magnesium deficiency and have been correlated with increased tendency toward bone demineralization, possibly mediated by both mechanisms: (1) lowering of alkaline phosphatase levels caused by magnesium deficiency, and (2) exceeding the capacity of the phosphatase available to destroy the excess phosphates.
Osteopenia, associated with hypophosphatasia, has developed in utero, as well as in infancy, childhood, and adult life (Rathbun, 1948; Sobel et al., 1953; Engfeldt and Zetterstrom, 1954; Schlesinger et al., 1955; Fraser, 1957; Currarino et al., 1957; Beisel et al., 1960; Lessell and Norton, 1964; Pourfar et al., 1972; Rudd et al., 1976). The most severe form is among those whose clinical manifestations develop earliest, possibly beginning in utero. Extensive osteopenic lesions that are found at birth, or in the early months of life, resemble those of osteogenesis imperfecta. Affected infants are assumed to have had spontaneous fractures that healed imperfectly and with angulation (Fraser, 1957). Similar fractures have been reported among infants vulnerable to prenatal and early infantile hypomagnesemia, particularly those born to preeclamptic women and to immature mothers with frequent or multiple pregnancies. Intrauterine growth retardation of abnormal pregnancies and placentas might give rise to fetal hypomagnesemia that can play a role in bone dysplasia. Possibly contributory is vitamin D administration during pregnancy, which has been shown to increase placental scarring in women. Hypervitaminosis D in pregnant rats has been shown to damage the placenta and has been implicated in the bone damage of the pups: thin bones with abnormal osteoid and spontaneous fractures. The lesions, like those of early severe hypophosphatasia, were considered similar to those of osteogenesis imperfecta, and were speculated to have been caused by passage of excessive vitamin D to the fetus through the damaged placenta (Ornoy et al., 1968, 1972). That excessive vitamin D can damage the osteogenic process, leading to lesions very much like those of severe early hypophosphatasia, was shown in 1932 by Shelling and Asher. Young rats on a diet that increased susceptibility to vitamin D toxicity (low in calcium and high in phosphate) showed progressive demineralization and replacement of trabeculae by osteoid remnants and tiny fragments of calcified material when they were given excessive vitamin D for 26 days. It is conceivable that fetuses of pregnant women who are hyperreactive to vitamin D, who consume excessive phosphate-containing foods and beverages, and who are magnesium deficient are at particular risk of developing bone dysplasia.
Possibly the unexplained convulsions of infants with early severe hypophosphatasia (Rathbun, 1948; Fraser et al., 1957; Currarino et al., 1957) might also be of hypomagnesemic derivation, such infants probably having poor skeletal magnesium reserves to meet the requirements of early life (especially in those who are fed cows' milk). The infants commonly suffer from irritability, anorexia, and persistent vomiting, and among those surviving to the second year, craniostenosis develops (Fraser, 1957). These manifestations again focus on the possible role of abnormal response to vitamin D as a contributory factor. They are comparable to those of infantile hypercalcemia, associated with hyperreactivity to vitamin D (Review: Seelig, 1969b), in which low levels of serum alkaline phosphatase have also been reported (Lightwood, 1932; Fanconi and Girardet, 1952; Schlesinger et al., 1956; Amann, 1959; Illig and Prader, 1959; Mitchell, 1960; Editorial, Lancet, 1960; O'Brien et al., 1960; N. David et al., 1962; Garcia et al., 1964; Fraser, 1966). Among 14 patients with hypercalcemia, not of hyperparathyroid origin, N. David et al. (1962) recorded low alkaline phosphatase in five with vitamin D toxicity or hyperreactivity. Another similarity of hypophosphatasia and hypervitaminosis D is the development of band keratopathy (Lessel and Norton, 1964) and chondrocalcinosis (O'Duffy, 1970) in hypophosphatasia and in vitamin D toxicity (J. E. Howard and Meyer, 1948; Chaplin a al., 1951, B. Andersen, 1956). In both hypophosphatasia and infantile hypercalcemia there is greater than normal susceptibility to vitamin D toxicity (Sobel et al., 1953; Reviews: Fraser, 1957; Seelig, 1969b), but the skeletal abnormalities of hypophosphatasia have not responded to vitamin D therapy (Engfeldt and Zetterstrom, 1954; Fraser, 1957). It is speculated that magnesium deficiency might underlie both the susceptibility to vitamin D toxicity, and the vitamin D resistance of the hypophosphatasia syndrome. Magnesium has been protective against development of cardiovascular and renal lesions of vitamin D toxicity. Yet, in magnesium deficiency there is vitamin D resistance.
In view of the fact that vitamin D excess causes magnesium depletion, it is of interest that chondrocalcinosis has also been reported in patients with hypomagnesemia and in experimental magnesium deficiency and phosphate loading, as well as in hypervitaminosis D (Christensen et al., 1951; DeWind, 1961). Vitamin D increases renal tubular reabsorption of phosphorus (Harrison and Harrison, 1941), as well as magnesium loss.
Nephrocalcinosis is common to hypophosphatasia (Review: Fraser, 1957), hypervitaminosis D (Review: Seelig, 1969b), and magnesium deficiency. The most notable difference between hypophosphatasia and infantile hypercalcemia is the osteopenia of the former and the osteosclerosis of the latter. It should be noted that vitamin D toxicity in animals on high intakes of calcium, and in children (most of whose vitamin D is in milk, which is rich both in calcium and phosphate), tend to have hypermineralized bones. Vitamin D and its metabolites, however, have bone mineral-mobilizing activity, and vitamin D toxicity in adults is generally associated with osteomalacia. High phosphate intakes are also implicated in osteopenia.
Several additional similarities to abnormal findings of magnesium deficiency have been reported in hypophosphatasia. The teeth are irregularly calcified and tend to be lost prematurely, a finding attributed to inadequate growth of alveolar bone (Fraser, 1957). Comparable changes have been described in magnesium-deficient rats (Bernick and Hungerford, 1965; Trowbridge and Seltzer, 1967) and hamsters (Yamane, 1962, 1970), and both spontaneously and experimentally in several species of animals when given diets high in phosphates.
Children with hypophosphatasia, whose lesions become apparent after the age of six months, generally have less severe bone lesions. They are characterized by coarse metaphyseal trabeculae that are distorted and irregularly calcified. Bernick and Hungerford (1965) described comparable lesions in magnesium-deficient rats. Possibly the two brothers with epiphyseal irregularities and areas of long bone rarefaction, who had hypercalcemia and hypophosphatasia, and were diagnosed as a rare form of renal rickets because of excessive renal tubular reabsorption of phosphorus (Schneider and Corcoran, 1950), might have had abnormal metabolism of magnesium, vitamin D, or both.
High urinary output of phosphoethanolamine is characteristic of patients with hypophosphatasia (Fraser, 1957). In view of the foregoing correlations of findings of this metabolic disorder, with some of those of magnesium deficiency, it is of interest that magnesium deficiency has caused urinary excretion of phosphoethanolamine in a third of the animals in which this parameter was explored, and that several magnesium-deficient patients also had both low serum alkaline phosphatase levels and high urinary outputs of phosphoethanolamine (Pimstone aet al., 1966). An unpublished observation of high phosphoethanolamine excretion in a woman with latent tetany of magnesium deficiency (Seelig et al., 1975) is of interest. In addition to excreting about 21/2 times more than normal amounts of phosphoethanolamine, she also had low alkaline phosphatase levels following a trial period of 25-OH-D3 therapy, during which her serum magnesium fell further. Her magnesium deficit has not been reparable because she is a renal magnesium waster (Seelig et al., 1976/1980).
12.6. Other Osteopenias Possibly Mediated by Magnesium Deficiency
12.6.1. Osteoporosis
There have been few studies on the influence of hormonal imbalances on bone magnesium accretion in postmenopausal osteoporosis, the most common cause of this disease. It has been estimated that no fewer than 6,000,000 have this disease in the United States (Harris and Heaney, 1969), even on the basis of the crude measure of osteoporosis provided by roentgenograms (which detects vertebral osteopenia only with loss of 30% to 50% of skeletal mass). The abnormalities in skeletal renewal that occur with metabolic bone disease and hormonal imbalances have been evaluated by Harris and Heaney (1969). Only those facets pertaining to possible mediating effects of magnesium loss in the hormonal imbalances are considered here. The available data suggest that magnesium loss from bone might contribute to several forms of osteoporosis.
Considering the effects of estrogen (and other female sex hormones), a deficiency of which has been most implicated in postmenopausal osteoporosis, and treatment with which has been and is under trial, there are fragmentary data that suggest that its effects on magnesium might be responsible for both promising and adverse effects. A clue to the retention of magnesium caused by estrogen was uncovered when analysis of metabolic studies showed that young women retain more of a marginal magnesium intake than do young men (Seelig, 1964). This observation was confirmed by Amiot et al. (1969) and Durlach (1970) in normal subjects and in patients with osteopenias. Comparable studies of magnesium retention of postmenopausal women have not been found. It was postulated that this difference in retention of magnesium might be a factor in the greater resistance of young women than men to cardiovascular disease (Seelig, 1964; Seelig and Lehr, 1971/ 1973; Seelig and Heggtveit, 1974), and might contribute to the increase in incidence of both cardiovascular and bone disease after the menopause (Seelig and Lehr, 1971/1973). On the other hand, plasma magnesium levels tend to be higher in young men than in young women, particularly during the period of greatest estrogen secretion, or when they are taking oral contraceptives (Dahl, 1950; N. Goldsmith, 1963; N. Goldsmith and Goldsmith, 1966; N. Goldsmith and Baumberger, 1967; DeJorge et al., 1967; Durlach, 1970; N. Goldsmith et al., 1970; Olatunbosun et al., 1974; 1976/1978; N. Goldsmith and Johnston, 1976/1980). Durlach (1970) cautioned that this effect of estrogens might contribute to thromboembolic phenomena, and recommended that women on oral contraceptives be given magnesium concomitantly to prevent increased coagulability that might be caused by lowered plasma magnesium levels. N. Goldsmith and Johnston (1976/1980) have reviewed the evidence as to the risk of thromboembolism in women on oral contraceptives.
Estrogen exerts both direct and indirect effects on bone metabolism. It inhibits bone resorption in vitro (P. Stern, 1969) and increases endosteal bone formation in mice (M. Silverberg and Silverberg, 1941), but decreases calcium accretion in rats, even though it decreases bone resorption (Lindquist et al., 1960). Estrogens antagonize PTH-induced bone resorption (Ranney, 1959; Nordin et al., 1970; Atkins et al., 1972), and in ovariectomized rats the bone-resorptive effect of PTH is increased (Orimo et al., 1972). Since PTH mobilizes bone mineral (including magnesium), estrogen has increased bone uptake of magnesium (N. Goldsmith and Baumberger, 1967), and magnesium deficiency causes osteopenia, it is possible that at least part of the effect of estrogen on bone might be mediated by its effect on bone magnesium levels. Another bit of evidence that implicates magnesium loss in some of the osteopenic processes is the degranulation of mast cells in magnesium deficiency (Hungerford and Karson, 1960; Bois, 1963), a process that causes release of heparin as well as histamine. Heparin enhances the resorptive response of bone to PTH (Goldhaber, 1965). Increased bone sensitivity to PTH has been implicated in osteoporosis, even in the absence of elevated endogenous PTH levels (Heaney, 1965; Harris and Heaney, 1969). Further support for this concept has been provided by Bélanger et al. (1975), who confirmed the damage to mast cells caused by magnesium deficiency, showed that female rats are more susceptible to magnesium deficiency-induced mast cell damage than are males, and that estradiol in the females and testosterone in the males resulted in less mast cell depletion.
Thus, there are data, deriving from magnesium-deficiency studies, that bear on some of the mechanisms that might be involved in the clinical benefit that has been reported with long-term prophylactic use of estrogens in postmenopausal women. Henneman and Wallach (1957) reviewed the records of 200 patients given estrogens by Albright and his colleagues for 1 to 20 years and found that, using loss of height as an index of osteoporosis, the use of estrogen arrested further loss of height in those already suffering from the disease, and prevented height loss in those whose postmenopausal estrogen treatment had begun before there was evidence of osteoporosis. [In regard to the concern about estrogen increase of cancer, the authors commented that in this group of 200 patients, who were given intermittent (cyclic) therapy, the incidence of carcinoma of the breast, cervix, and endometrium was low.] Determination of the effect of estrogens on bone thickness by means of densitometry has also shown estrogens to inhibit progression of postmenopausal osteoporosis (Meema and Meema, 1968; M. E. Davis et al., 1966; Meema et al., 1975; N. F. Goldsmith and Johnston, 1975,1976/1980). Estrogen has also been shown to decrease bone resorption, as measured by urinary output of hydroxyproline (Riggs et al., 1969; Gallagher and Nordin, 1972) and to be effective (in doses of no less than 1.25 mg of conjugated estrogen in a series of 220 severely osteoporotic women) in arresting vertebral fractures (Gordan, 1971). The mechanism of action has been postulated to be via estrogen inhibition of PTH-induced bone resorption in post- menopausal women (Nordin, 1971; Gallagher and Nordin, 1972; Seelig and Lehr, 1971/1973).
Why women are more susceptible than are men, in the middle years, to (presumed) relative hyperparathyroidism is not clear. It is possible that the estrogen-induced lowering of plasma magnesium (which might be the result of a shift to intracellular sites) might result in chronic stimulation of parathyroid secretion. If such stimulation causes parathyroid hyperplasia [as Larvor et al. (1964a) have demonstrated in calves], when the ovaries cease functioning the overactive parathyroids might continue to mobilize bone minerals, excessively in the absence of the counteracting effect of estrogen (Fig. 12-6).
The later development of osteoporosis in men probably reflects their longer gonadal activity. Testosterone has also been shown to have activity in clinical osteoporosis (Gordan, 1954).
Evidence that calcitonin (CT) retards disuse osteoporosis (Hantman et al., 1973) and that magnesium administration stimulates CT secretion suggests that magnesium administration may be useful in this form of osteoporosis. It recalls the work with rats, showing interrelationships among magnesium, CT, PTH, and cortisone (Palmieri et al., 1969; Eliel et al., 1971). Cortisone, an excess of which has long been known to cause osteoporosis, abolished the hypomagnesemic effect of CT, an effect attributed to its interference with CT inhibition of bone-resorption. On the other hand, patients with regional enteritis had their magnesium deficiency (to the point of hypomagnesemia) intensified by corticosteroid therapy (Gerlach et al., 1970). Although only the acute signs of magnesium deficiency were considered in that paper, it should be recalled that malabsorption is implicated in osteopenia (i.e., celiac rickets and osteomalacia), as are corticosteroid therapy and magnesium depletion.
Administration of magnesium supplements to several patients, including a few with conditions (e.g., alcoholism or cirrhosis) that predispose to magnesium deficiency, improved their calcium retention (Briscoe and Ragan, 1966). Du Ruisseau and Marineau (1971/1973) showed that patients with osteopenia retained more calcium when supplemented with magnesium. In contrast, administration of calcium to such patients increased their magnesium deficit (Table 12-4, Fig. 12-7, Parlier et al.,. 1963; Amiot et al., 1969).
12.6.2. Renal Osteodystrophy
The osteopenia seen in patients with uremia, whether they are dialyzed or not, entails complex interrelationships among etiologic and complicating factors; space does not permit their consideration here. Selected from the massive literature on this subject are data directly or indirectly bearing on the possibility that tissue magnesium depletion might play a role. Serum magnesium levels are unreliable as an index of the magnesium status of uremic patients. High normal, and low levels have been reported that are unrelated to tissue levels (Lim et al., 1969a; Lim and Jacob, 1972c). Metabolic balance determinations are also unreliable, the equilibrium reported by Clarkson et al. (1965) being associated with subnormal intestinal magnesium absorption that balanced its subnormal urinary excretion. In contrast, patients with chronic renal failure receiving low protein diets, lost as much as 139 mg of magnesium daily, despite slightly elevated serum magnesium levels (Kopple and Coburn, 1973).
Massry and Coburn (1970a) proposed that tissue magnesium deficiency in patients with progressive renal failure (even when serum magnesium levels are elevated) might be contributory to their hypocalcemia, vitamin D resistance, and defective response of the skeleton to parathyroid hormone. On the other hand, if the tissue magnesium deficit involves the parathyroids, secondary hyperparathyroidism might ensue. Pletka et al. (1971) have, in fact, shown that the levels of parathyroid hormone (PTH) of patients treated by hemodialysis with water containing 1.5 to 2.5 mEq of magnesium per liter fell 20% from pretreatment high levels. Those who were hemodialyzed with water containing low concentrations of magnesium (0.5 mEq/liter), who had lesser initial elevations of PTH, showed a 118% rise in PTH after two months of treatment. It is well known that patients being treated by dialysis are subject to hyperparathyroidism, with the attendant problems of bone loss and metastatic calcification (Buckle, 1970; Kleeman et al., 1970; Genuth et al., 1970; Danesh et al., 1970; Terman et al., 1971; Henderson et al., 1971; Editorial, Brit. Med. J., 1972; Arora et al., 1975). Cardiovascular involvement is common, and cardiovascular disease is by far the leading cause of death in patients on chronic dialysis (Lowrie et al., 1974), accelerated arteriosclerosis (Lindner et al., 1974; Curry and Roberts, 1977), myocardial calcification, and heart block having been reported.
There is concern about producing hypercalcemia and possibly hypermagnesemia by using untreated hard water (Freeman et al., 1967). Acute symptoms of hypermagnesemia (blurred vision, flushed face, weakness, and inability to stand) were produced by use of a dialysate containing 15 mEq of magnesium per liter (Govan et al., 1968). Posen and Kaye (1967) have reported that magnesium levels in the dialysis bath water in major centers range from almost 0 to 0.8 mEq/liter, usually depending on the concentration in the water supply; although they used Montreal water (one of the harder water supplies available), they added 1 mEq of magnesium to each liter, so as to provide 1.65 mEq/liter. They attribute to the added magnesium the freedom of their patients from metastatic calcification over the four-year observation period. They did not comment on the incidence of osteodystrophy, but Catto et al. (1973) commented that osteodystrophy is not a problem in Montreal or London (both hard-water cities), whereas it is in Newcastle and Los Angeles. Kleeman et al. (1970), who commented on the magnesium supplied by two medical centers in Los Angeles, 0.5 and 1.5 mEq/liter, suggested that providing a dialysate magnesium concentration (1.5 mEq/liter) sufficient to prevent hypomagnesemia might reduce the tendency toward metastatic calcification and secondary hyperparathyroidism. This recommendation was also made by Danesh et al. (1970). It may be relevant that the accelerated arteriosclerosis reported by Lindner et al. (1974) was from a center in Seattle, a soft-water area. The low incidence of osteodystrophy in two hard-water cities (Catto et al., 1973) suggests that the magnesium provided might also protect against secondary hyperparathyroidism and osteodystrophy.
However, there is no consensus as to the optimal magnesium concentration of water used for dialysis. Unlike Posen and Kaye (1967), who added magnesium to the hard water, it is common to attempt to bring the serum magnesium levels down to normal limits by using dialysates with low magnesium concentrations (from 0.16 to 1.0 mEq/liter) (Johny et al., 1971; W. K. Stewart and Fleming, 1971; 1973; Paschen et al., 1971). In one reported patient receiving twice-weekly hemodialysis with water containing 0.8 mEq/liter of magnesium, severe hypomagnesemic cramps developed that promptly responded to magnesium therapy (Triger and Joekes, 1969). In view of the cited evidence that tissue levels of magnesium can be low in patients with renal disease, despite high serum levels, and the importance of tissue magnesium in protecting against pathologic changes in cardiovascular and skeletal tissues, the advisability of depleting the body magnesium by use of low magnesium dialysate is open to question.
Since bone and serum magnesium tend to be in equilibrium, the fact that surface bone magnesium levels of uremic patients tend to be higher than normal (Contiguglia et al., 1972; A and Miller, 1973) is not surprising. Its significance is uncertain. Alfrey and Miller (1973) found that 30% of the bone magnesium of uremic patients with hypermagnesemia is within the bone hydration shell or on the crystal surface, and speculate that, since magnesium can influence crystal size and stability, an excess might play a role in osteodystrophy. They noted, however, that the deeper magnesium is not as readily exchanged, and its mobilization is dependent on the resorptive process. However, chronic experimental uremia adversely influences collagen metabolism in both skin and bone (Hahn and Avioli, 1970). Also, experimental magnesium depletion causes formation of abnormal bone matrix with defective calcification capacity. Thus, it seems likely that loss of deep-located bone magnesium should have a more significant effect on the osteopenia of renal disease than the gain at the surface.
A final indirect bit of evidence that magnesium deficiency might contribute to renal dystrophy is the observation that renal osteodystrophy is rare in Israel (Berlyne et al., 1973b). The rarity of this disease (in Beer Sheva) was attributed by the investigators to the low phosphorus intake of dwellers in that area. However, in another publication, Berlyne et al. (1973a) reported that the water in that area was also very high in magnesium and calcium.
12.7. Joint Diseases Possibly Mediated by Magnesium Deficiency
12.7.1. Osteochondrosis
There are meager clinical data that suggest that magnesium deficiency might play a role in osteochondrosis or osteochondritis (Legg-Perthes disease; slipped epiphyses). J. F. Miller (1944) reported a child who had had neonatal tetany and hyperirritability and cyanotic episodes during the early weeks of life, for which he was given calcium therapy, which was continued (with halibut liver oil) from then on. By 6 months of life he developed normocalcemic convulsions that stopped at the age of one; his tremors persisted. At 31/2 years of age he had osteochondrosis of the capital epiphysis of the left femur, including fragmentation and flattening of the epiphysis. At that time he had hypercalcemia (12.9 mg/100 ml). By the age of 6 years, in addition to dizziness and tremors, he had developed muscle cramps and carpopedal spasms, at which time his plasma magnesium was determined for the first time; it was 1.4 mEq/liter. He responded strikingly to magnesium therapy (300 mg MgSO4 three times daily), with disappearance of tremors and dizziness. After the supplements were stopped by his parents for a week when he suffered an attack of bacillary dysentery, his tremors and dizziness recurred, he had positive Trousseau and Chvostek signs, and his plasma magnesium dropped to 0.5 mEq/liter. He again improved promptly on magnesium therapy. The osteochondrosis had been treated surgically, and thus the effect of the magnesium therapy on this disease could not be ascertained, but Miller speculated that there might have been a relationship between the boy's probable early magnesium deficiency and his epiphyseal abnormality. Klingberg (1970) reported mild osteochondritis of shoulders, knees, and hips (Legg-Perthes-like) in a boy who suddenly developed a carpopedal spasm of 6 hours duration at 5 years of age and who was found to have both hypomagnesemia (0.8 mEq/liter) and hypokalemia (2.8 mEq/liter). A 6-day metabolic balance study showed minimal negative magnesium balance; supplementation with 60 mEq magnesium (as the acetate) produced a slightly positive magnesium balance (+42 mEq) he continued to excrete 5-9 mEq/day in his urine. After 6 months of magnesium supplementation, the patient's bony lesions reverted to almost normal. With the lower magnesium supplements, his tetany recurred. The possibility of a renal tubular defect in magnesium reabsorption was proposed as an explanation of the child's high magnesium requirement. Follow-up of this child for 6 years has shown persistence of his renal wastage of magnesium. He has also developed cardiac and skeletal abnormalities (W. O. Klingberg, personal communication, 1978). Children with hypophosphatasia (proposed as related to magnesium deficiency) also have metaphyseal and epiphyseal abnormalities, as have some children with vitamin-D refractory rickets, also related to magnesium deficiency.
The studies that show abnormalities of metaphyseal trabeculae and of the epiphyseal cartilage and ground substance (Yamane, 1962; 1970; Yamane and Singer, 1953; Bernick and Hungerford, 1965; Clark and Bélanger, 1967; Trowbridge and Seltzer, 1967; Trowbridge, 1971) provide direct evidence that experimental magnesium deficiency causes abnormalities in epiphyseal structure. Abnormal epiphyseal cartilage and diaphyses have also been seen in pups of pregnant rats overdosed with vitamin D (Ornoy et al., 1972), and cessation of epiphyseal osteogenesis in young rats with vitamin D toxicity (Shelling and Asher, 1932; Ham and Lewis, 1934; Storey, 1960), lesions that might reflect secondary magnesium deficiency.
Before a generalization can be drawn (correlating clinical epiphyseal disease with early magnesium deficiency), there should be evaluation of children with this disease, and of their families, for abnormalities in magnesium absorption and retention.
12.7.2. Chondrocalcinosis and Osteoarthritis
Enlargement of the joints and marked stiffness were identified as signs of magnesium depletion in calves and were shown to resolve when magnesium salts were added to a high-phosphate, low-calcium diet by Huffman et al. (1930). Deletion of the magnesium carbonate supplement resulted in recurrence of the stiffness within two months. Cattle foraging in low-magnesium areas also developed articular damage, in these instances characterized by erosions of the cartilage (Willers et al., 1965). House and Hogan (1955) demonstrated that optimal intakes of magnesium (0.35% of diet) and potassium (1.5% of diet) prevented the stiffness and periarticular deposition of calcium phosphate that developed in magnesium-deficient guinea pigs receiving only slightly more phosphorus than calcium (P/Ca = 0.9/0.8%) (Hogan et al, 1950). Joint stiffness was worst in guinea pigs fed rations containing 1.7% phosphorus, 0.9% calcium, 0.04% magnesium, and 0.41% potassium.
Chondrocalcinosis has also been associated with human diseases associated with magnesium loss. The first instances were in rheumatoid arthritis patients taking excessive amounts of vitamin D (Review: Christensen et al., 1951). Additional to the metastatic calcification of the arteries, kidneys, and other viscera, there was sometimes marked and disabling calcification of the periarticular structures, involving the synovial cavities, bursae, and tendon sheaths (accompanying generalized osteoporosis). Withdrawal of the toxic vitamin D supplements resulted in decreased periarticular calcification. This condition was usually seen among rheumatoid arthritis patients who had self-medicated themselves with vitamin D supplements providing as much as 200,000 units daily. It was also seen in a child who had been given high dosage (> 40,000 units/day) vitamin D since the age of three because of suspected rickets, diagnosed on the basis of a "peculiar feeling to the skull," as well as wrist changes. When seen by the investigator (DeWind, 1961) at 5 he had periarticular calcification, as well as osteosclerosis that encroached on the medullary canals. The bone changes resemble those described in experimental magnesium deficiency.
A patient with monoarticular osteoarthritis, whose hypophosphatasia was diagnosed in middle age, had calcification of the articular cartilage of her hips, symphysis, and arthritic knee (O'Duffy, 1970). This was the first time note was taken of the deposition of calcium pyrophosphate in cartilage of a patient with hypophosphatasia, but O'Duffy reviewed the literature and found several additional cases in which periarticular calcification was noted in the case reports. He reviewed some of the metabolic disorders in which pseudogout was reported, and found that it was common in hyperparathyroidism. McCarty et al. (1974), who compared the frequency of concomitant chronic diseases in patients with pseudogout and in those with osteoarthritis of the large weight-bearing joints, found no significant differences, and that immunoreactive parathyroid hormone was elevated in both groups. They postulate that sustained low-grade hyperparathyroidism might be related to the genesis of the articular lesions. This is a provocative observation, since both vitamin D excess and hyperparathyroidism are associated with loss of magnesium. McCarty (1974) and his co-workers (McCarty et al., 1971) have related magnesium with inhibition of calcium pyrophosphate precipitation in synovial fluid, correlating this effect with magnesium-activation of pyrophosphate transphosphorylase.
Precipitation of calcium pyrophosphate in the joints of patients with hypomagnesemia has been reported. McCarty et al. (1974) reported one such instance in a patient with psoriasis. Runeberg et al. (1975) reported a young man who had renal tubular magnesium wasting, hypomagnesemia, and from whose knee joint calcium pyrophosphate crystals were obtained. This patient is of particular interest, since he had had nephrocalcinosis from the age of seven, following calcium therapy of his convulsive hypocalcemia, and developed ECG changes similar to those seen with magnesium depletion when he was 14 years of age. After he retained 247 mmol of magnesium (during a period of intravenous infusions of 4 mmol of magnesium chloride daily for 8 days), he was maintained on high oral magnesium dosage (20mmol as Mg (OH)2 and 30 mmol as MgCl2 and potassium for 2 years. His joint effusion disappeared and he remained symptom-free since. Rapado and Castrillo (1976/1980b) reported a man of 38 with knee joint pain and swelling of several years duration, who also had renal tubular magnesium wasting and hypomagnesemia. He had X-ray evidence of linear calcification of the cartilage, and biochemical demonstration of calcium pyrophosphate in a synovial biopsy. This patient, too, responded to magnesium therapy, but his response is not as clear-cut because he was maintained also on antiinflammatory drug therapy
Ankylosing hyperostosis, a common disorder of the middle-aged and elderly that affects the spine and large joints, has also caused calcaneal spurs (particularly of the heel), and has also been associated with precipitation of crystals of calcium pyrophosphate dihydrate. Among 30 patients reported by Utsinger et al. (1976), one had hypomagnesemia, three had hyperphosphatemia, and four had elevated serum alkaline phosphatase. More intensive study of the magnesium status should yield useful data.
Complicating the problem of chondrocalcinosis and exostosis and their response to magnesium is pyrophosphate's inhibition of precipitation of calcium phosphate compounds in urine (Fleisch and Neuman, 1961; Fleisch and Bisaz, l962a), and the necessity of pyrophosphatase for normal (bone) mineralization. Thus, there must be a delicate balance between enzymes and substrate on the one hand, and concentrations of the minerals: magnesium, calcium, and phosphorus on the other. Hydroxyapatite crystals are most commonly found in periarticular disease, in contrast to the calcium pyrophosphate dihydrate that is more frequent in intraarticular disease, such as is not uncommon in uremia (Parfitt, 1969). Mirahmadi et al. (1973) reported that calcium hydroxyapatite has precipitated periarticularly in renal failure patients undergoing hemodialysis: They suspect that hyperphosphatemia is the most likely provocative factor. They noted that none of their seven patients with this complication had magnesium depletion, and recommended measures to lower the serum phosphate levels and use of higher calcium concentrations in the dialysate, to suppress parathyroid secretion. Since increased magnesium also suppresses the parathyroid function, further study and individualization of the prophylactic or therapeutic regimen is advisable.
Ankylosing spondylitis, accompanying bone resorption (from adolescence on), and irregularity and erosion of the articular cartilage (such as has been reported in magnesium deficiency, supra vide), with obliteration of the joint space, has been encountered in primary hyperparathyroidism (Bunch and Hunder, 1973), a condition associated with magnesium loss.
Articular lesions-peri-, para-, and intraarticular calcification-have also been seen in uremic patients (Review: Parfitt, 1969). An unusual paraarticular lesion occasionally seen in such patients, and that had been more common when high doses of vitamin D were given as treatment for arthritis (Christensen et al., 1951) is tumoral calcinosis, rubbery or cystic calcific mass. McPhaul and Engel (1961) reported two patients with this disorder in one family, four of whom-including the patients-had low plasma alkaline phosphatase levels. Parfitt (1969), who reviewed the factors involved in soft tissue calcinosis of uremia (including the articular forms calcification), considers hyperphosphatemia the most important single factor, and calciphylaxis (produced by prior "sensitization" with vitamin D or parathyroid hormone) as a unifying hypothesis. Since Selye, who promulgated the calciphylaxis theory, found magnesium to be protective (against cardiorenal calcinosis caused by high phosphate, vitamin D, or PTH), perhaps low magnesium levels caused by these agents (as well as by uremic acidosis) might play a role in the abnormal calcification processes of the joints in uremic patients. Whether magnesium deficiency also plays a role in osteoarthritis cannot be averred in the absence of other than the meager animal and clinical data available.
Leonard and Scullin (1969) and Leonard et al. (1971) have proposed that in soft tissue, where the concentration of magnesium exceeds that of calcium, the formation of MgATP inhibits calcium apatite formation. This group has demonstrated that the local magnesium/calcium ratio influences calcification of tendons (in turkeys) and that egress of magnesium precedes the onset of calcification (Leonard et al., 1976). This is a physiologic maturation process in turkey tendon. It seems plausib1e that low Mg/Ca concentrations in soft tissues, and in articular and periarticular tissues, might similarly participate in calcification, and that a higher magnesium concentration might inhibit it.
12.8. Magnesium Deficiency and Dental Disorders
Damage to teeth, as well as to bones and to soft tissues, were among the findings reported from the earliest magnesium deficiency studies. Kruse et al. (1932) found that rats surviving severe magnesium deficiency for 10 weeks had loose molars and incisors. Further data on the abnormal periodontal soft tissues were provided by H. Klein et al. (1935). Brittle, chalky teeth (loose in their sockets) were noted by Watchorn and McCance (1937) in subacute magnesium-deficient rats. They, like H. Klein et al. (1935), found striations in the dentin, suggestive of intermittent interference with the calcification process; they also reported odontoblastic degeneration. Becks and Furata (1939, 1941) reported pronounced degenerative changes in the enamel epithelium of rats by the 72nd day of subacute magnesium deficiency. Irving (1940) confirmed the damage to the enamel, caused by magnesium deficiency, as well as striations in the dentin. He also noted increased width of the predentin above the basal portion of the teeth that he considered unique to magnesium deficiency. Yamane and Singer (1953) found alternate bands in the incisors of magnesium-deficient hamsters that were associated with odontoblastic degenerative changes, and decreased width of the enamel-forming cells (Yamane, 1962). Bernick and Hungerford (1965) showed that magnesium-deficient rats had disturbed dentin calcification. It was characterized by a wide uncalcified zone separated from the predentin by a thin calcified line. There was also odontoblastic degeneration. Trowbridge (1971) and Trowbridge et al. (1971) point out that magnesium deficiency also causes dentinal striations and that the incisal dentinal striations ceased within four days of magnesium supplementation; thereafter the new dentin was normal except in areas adjacent to enamel, where it was somewhat attenuated.
The importance of magnesium for the metabolism of teeth is suggested by the avidity of teeth of control magnesium-deficient lambs for 28Mg (McAleese et al., 1961). F. R. Morris and O'Dell (1961) had shown that increasing the phosphorus content of the diet from 0.4 to 1.7% did not affect the calcium or phosphorus content of the teeth but intensified magnesium deficiency, which they had earlier shown to cause formation of the defective teeth and decay; the teeth were loose in their sockets (O'Dell et al., 1960). The authors commented that their findings suggested that the magnesium deficiency probably affected cell function and development of the organic matrix of the tooth, rather than its mineralization. That the organic matrix of dentin of magnesium-deficient rats did, indeed, differ from that of controls was demonstrated by Bernick and Hungerford (1965). Differences in staining characteristics suggested that the ground substance of the matrix of bones and teeth of magnesium-deficient rats contained less polymerized mucopolysaccharides; they are thus less subject to normal calcification. Defective dentin matrix formation by acutely magnesium-deficient rats was confirmed by Trowbridge and Seltzer (1967). They demonstrated greatly reduced tritiated proline labeling in the organic matrix of the dentin and retarded dentin formation and calcification, arrested appositional bone growth and resorption of the crest of the alveolar process (Trowbridge and Jenks, 1968; Trowbridge, 1971). The periodontal ligament was wider in the magnesium-deficient rats than in the controls, and there was minimal osteoblastic activity and lesser evidence of alkaline phosphatase activity in the pulps and the serum of the deficient rats. Magnesium-deficient hamsters also had periodontal abnormalities, as compared with pair-fed controls (Yamane, 1962, 1970). The periodontal ligament was disorganized, calculi formed in the gingival sulci, and the interdental bony septum showed resorption. Following extraction of teeth, the magnesium-deficient hamsters exhibited delayed healing, an effect attributed to impaired matrix formation. Delayed eruption of the permanent teeth, as well as abnormal mineralization of both dentin and enamel, odontoblastic degeneration, arteriosclerosis of pulpal vessels, and pulpal calcification were reported by Binus (1968) in magnesium- deficient dogs.
Two genetic clinical abnormalities that the author postulates may be associated with magnesium depletion: hypophosphatasia (Reviews: Fraser, 1957; Currarino et al., 1957) and pseudohypoparathyroidism (Review: Bronsky et al., 1958) are associated with dental disorders that bear some resemblance to those of experimental magnesium deficiency. In hypophosphatasia irregular calcification and severe caries have been reported. Three quarters of the children whose disease became manifest by the sixth month of life had premature loss of teeth, attributed to inadequate growth of alveolar bone and incomplete formation and early resorption of the roots of the teeth (Review: Fraser, 1957; Pourfar et al., 1972). Beisel et al. (1960) reported early loss of all permanent teeth of a patient who presented his first signs of hypophosphatasia as an adult. Both in pseudohypoparathyroidism and in idiopathic hypoparathyroidism, comparable dental abnormalities are not uncommon (Table 12-5) (Bronsky et al., 1958).
It is noteworthy that in two conditions with abnormal vitamin D metabolism, dental abnormalities have been reported. Children with hyperreactivity to vitamin D, who also have hypophosphatasia, have a high incidence of malocclusion, enamel hypoplasia, and severe caries. Rampant caries, necessitating early extraction of all teeth (before the age of 20) has been reported in a patient with familial hypophosphatemic vitamin-D-resistant rickets (Blackard et al. 1962). Enamel hypoplasia involving teeth that calcify after birth was found in members of a family with hypophosphatemic rickets and secondary hyperparathyroidism (Arnaud et al., 1970). On the other hand, periodontal disease has been correlated with high phosphate intakes and secondary hyperparathyroidism and with osteoporosis (Lutwak, 1974; Review: Krook et al., 1975).
(include the word "jacket" to search only in this book)
| Jacket | Preface | Contents | Introduction (Chapter 1) |
Chapter: | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| Appendix | Bibliography (A-D), (E-K),
(L-R), (S-Z) |
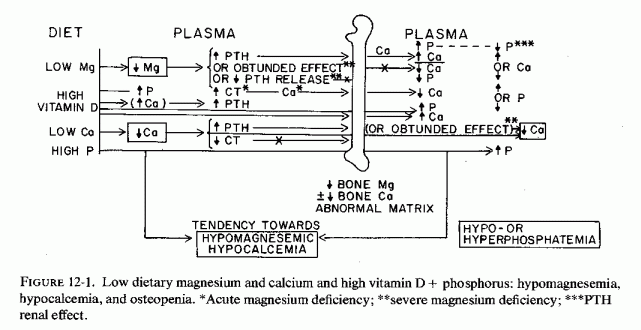{kind=link}
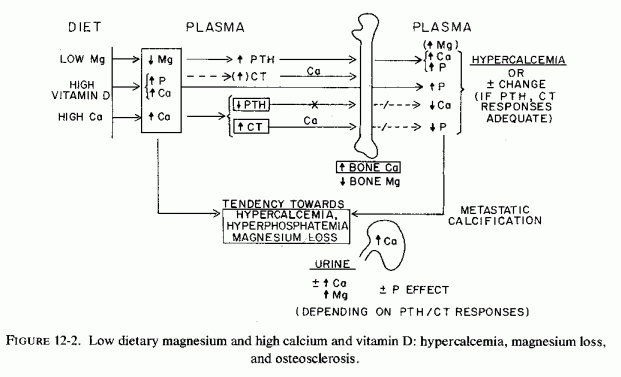{kind=link}
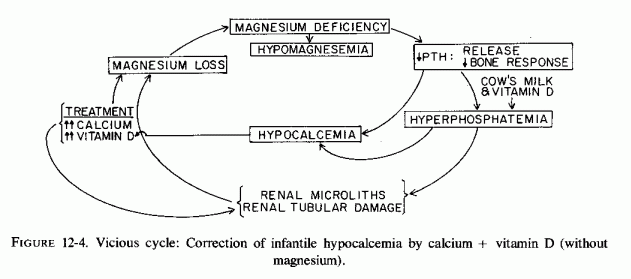{kind=link}
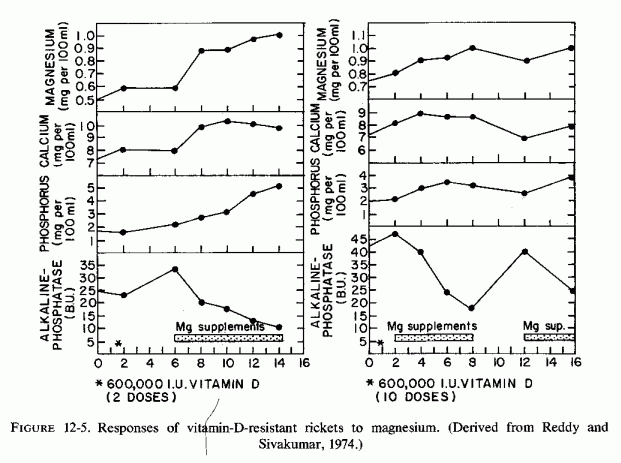{kind=link}
{kind=link}
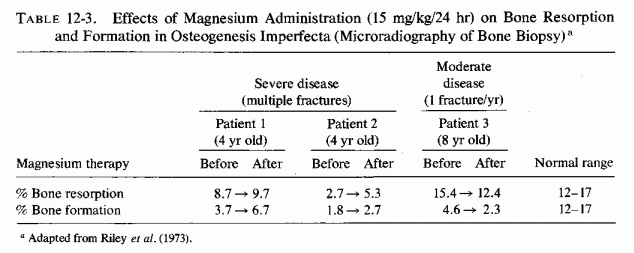{kind=link}
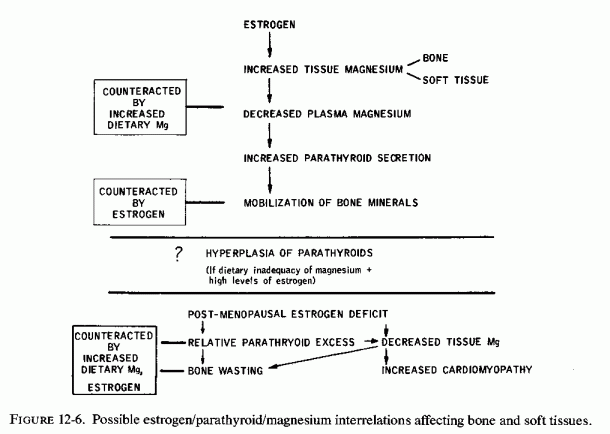{kind=link}
{kind=link}
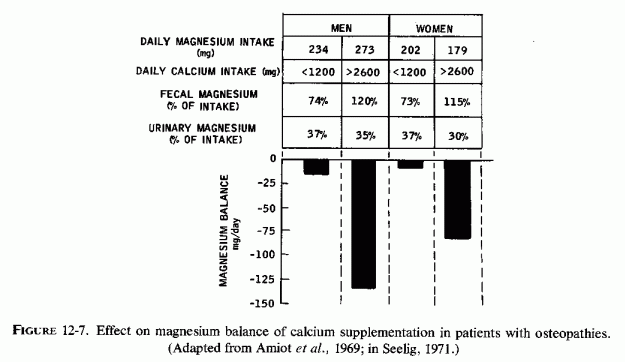{kind=link}